This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
FDA/CBER (the Agency) provided an overview of CMC challenges for CART development (e.g., Use of automation does not allow for bypassing FDA requirements with respect to method transfer comparability. Opportunity for feedback from FDA if provided pre-BLA. Release testing alone is not sufficient.
With the corrective and preventive action (CAPA) process often a “one-size-fits-all” approach, a whitepaper by the Medical Device Innovation Consortium (MDIC) Case for Quality Collaborative Community (CfQcc) has instead shared a risk-based framework focusing on continuous improvement. These results are included in most recent paper.
This is also evident from the number of US Food and Drug Administration (FDA) approvals and European Medicines Agency (EMA) approvals, which still show a large number of approvals on the small molecule side. I think we are increasingly seeing the focus move to globally approvable drug substances or drug products.
FDA and EMA Updates: Regularly monitor guidelines from agencies like the FDA (U.S.) Technologies like AI, big data, and AR/VR streamline processes, improve targeting, and create more engaging campaigns. Navigating the legal landscape requires agility and foresight, especially when new regulations surface.
Since 2016, drug approvals per clinical campaign have outpaced the average across all modalities, resulting in 11 FDA-approved therapies in that span. Combining the incremental processimprovements previously mentioned results in an optimized facility, with the adjusted block flow shown in Figure 5.
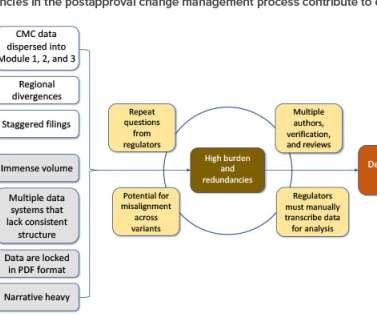
Application of this innovative approach will quickly orient regulators to the content of Module 3, “present product quality benefit-risk considerations, summarize the pharmaceutical development, present an overall understanding of the product quality,” 1 and facilitate continuous improvement.
Application of this innovative approach will quickly orient regulators to the content of Module 3, “present product quality benefit-risk considerations, summarize the pharmaceutical development, present an overall understanding of the product quality,” 1 and facilitate continuous improvement.
Recently, the European Medicines Agency (EMA) took the lead in pushing for processimprovements using technologies already established in other manufacturing sectors. Despite this, only 13 nanomedicines had been approved by the US Food and Drug Administration (FDA) before 2015. Yet in 2021, 100 nanomedicines had been marketed.
MyMD plans to present the data to the US Food and Drug Administration (FDA) and intends to advance the clinical programme for MYMD-1 into Phase III either alone or through partnership. There are no FDA-approved medications to treat sarcopenia. Why is a TNF-α inhibitor promising as a first-in-class therapeutic for sarcopenia/frailty?
Following approval of an initial marketing application, postapproval changes are needed to ensure adequate supply, mitigate supply risk, expand patient market access, optimize manufacturing processes, improve analytical methods, and comply with new regulatory expectations. Pharmaceutical Engineering 41, no. Draft Guidance for Industry.
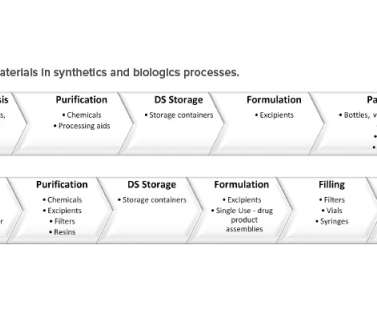
In addition, single-use technologies have been increasingly employed throughout manufacturing because of the advantages they offer, including reductions in cost, manufacturing footprint, contamination risk, and processing times (Figure 1). There is undoubtedly a need for improved supply chain flexibility to address shortages.
FDA Official Warns Manufacturers of Common Problems Found in Aseptic Operations.” www.raps.org/news-and-articles/news-articles/2023/3/fda-official-warns-manufacturers-of-common-problem 25 US Food and Drug Administration Center for Drug Evaluation and Research. September 2022. 25%20from%202022%20to%202027 24 Eglovitch, J. 7 March 2023.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.

















Let's personalize your content