This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
A study of the causes of warning letters issued by the US Food and Drug Administration (FDA)’s Center for Drug Evaluation and Research (CDER) and Center for Devices and Radiological Health (CDRH) between 2010 and 2020 revealed that poor current good manufacturing practice (cGMP) compliance and misbranding were the most common citations.
Draft guidance published by the US Food and Drug Administration (FDA) in December 2023, discussed quality considerations for topical ophthalmic drug products, including key considerations for extractables and leachables (E&L) testing. This document is revised from a version published in October 2023.
Food and Drug Administration (FDA) issued two guidance documents, one draft and one final, on food allergen labeling requirements. In addition to providing additional examples of tree nuts, the draft guidance states that FDA considers the following categories of fish to be major food allergens under Section 201(qq): Jawless fish (e.g.,
The rules were implemented to enhance patient safety protections via revised drug handling, packaging, and delivery requirements. The rules include changes to the notification process, medication packaging, the handling of reports, and safety issues.
After a firm submits a 510(k) to FDA, FDA will request still more information after a first-pass review. According to the 2 nd Quarter FY2023 MDUFA V Performance Report , FDA issued a request for additional information (AI request) on the first FDA review cycle for 63% to 68% of 510(k)s submitted in FY2018 to FY2022.
Packaging plays a critical part in the pharmaceuticals and medical devices industries and is developed with its own set of security standards for the safety of consumers. The role of primary packaging has extended beyond the primary objectives of sterility, physical and chemical protection, and security.
Mullen — More than five years after FDA first announced its plan to harmonize 21 CFR Part 820 with ISO 13485, on February 2, 2024, FDA finally issued the Quality Management System Regulation (QMSR) Final Rule. FDA further retained some definitions in the QSMR. Notably, Part 820 will look different. The new § 820.10
Livornese — FDA issued a Federal Register notice on September 13, 2024, seeking feedback on the Integrated Review format that the Center for Drug Evaluation and Research (CDER) began using as part of its New Drugs Regulatory Program (NDRP) modernization effort several years ago. By Deborah L. 89 FR 74966 at 74968 (Sept.
The Washington Nationals won the World Series, Presidential administrations have come and gone, and FDA has added new meeting types and formats to its menu. And so, FDA has issued a new draft guidance to bring everyone up to speed on formal meetings under PDUFA. Tobolowsky — Much has changed since the long-gone days of 2017.
The sampling strategy must be supported by sound and properly cited sources whose conclusions must be presented as supportive elements in the study documents. The following four elements of sampling methodology were found to be under-documented in RMM effectiveness studies: Supporting documentation for country/region selection.
There are common, though preventable, pitfalls that applicants may encounter, including incomplete or inadequate nonclinical data, which can be addressed by ensuring that the package of nonclinical studies submitted in the application adhere to applicable ICH and FDA guidelines.
Koblitz — FDA uses its Product Specific Guidance documents (“PSGs”) to provide recommendations as to the bioequivalence testing necessary for approval of a generic drug. FDA may deny a PSG if the applicant’s bioequivalence testing started after the PSG publication.
Food and Drug Administration – labeled an “Official Action Indicated” classification – is generally devastating for the facility, not least because it can stall FDA approval of applications to market drugs manufactured at the facility. And the Guidance may be used as leverage to secure action from FDA on a meeting request.
Such codes need to be placed on device labels and packages to allow devices to be easily identified and tracked throughout their lifecycle, except where the rule provided for an exception or alternative. The compliance dates were first published in 2013, and subsequently updated in various guidance documents and regulations published by FDA.
By Riëtte van Laack — On September 28, 2022, FDA announced the availability of the proposed rule for the implied nutrient content claim “healthy.” The term healthy, as an implied nutrient claim, was first defined by FDA in 1994. FDA also announced it would be re-evaluating the regulatory criteria for use of the “healthy” claim.
Brevig, Senior Regulatory Device and Biologics Exper — FDA recently published Alternative or Streamlined Mechanisms for Complying with the Current Good Manufacturing Practice Requirements for Combination Products; List under the 21st Century Cures Act in the Federal Register (FR Notice). FDA must also review the list periodically.
Earlier in March, the FDA shared a draft guidance on how to run clinical trials for the accelerated approval of cancer drugs. There, the FDA suggested that the drugs undergo randomised controlled trials, which the document describes as the preferred approach, rather than single-arm trials.
Over the next year, IMPACT authored the following documents for the client: Pre-NDA meeting package. Working with the company’s electronic publishing vendor to ensure that fully-compiled, submission-ready documents were produced. 5 clinical study reports. and 2.7.4), and the Clinical Overview (Module 2.5).
Over the next year, IMPACT authored the following documents for the client: Pre-NDA meeting package. Working with the company’s electronic publishing vendor to ensure that fully-compiled, submission-ready documents were produced. 5 clinical study reports. and 2.7.4), and the Clinical Overview (Module 2.5).
This tracking involves the use of unique product identifiers, such as serial numbers and barcodes, on drug packaging to track their movement. This record-keeping includes detailed documentation of the product’s history and movement through the supply chain. It also enables precise tracking and tracing of each drug package.
Small Dispensers and the 6/12/2024 FDA Announcement Concerning DSCSA This week’s article on the recent DSCSA postponement comes from PRS Pharmacy Services. If the FDA or another regulator requests DSCSA transaction data, the DSCSA regulations give you up to 48 hours to respond. The original article can be found HERE.
Improvements have been observed by various regulatory agencies in Latin America for the acceptance and implementation of international standards—for instance, the ICH Common Technical Document (CTD) format. Additional required information beyond ICH guidelines, or non-value-added documents (e.g., 6, and 2.3.S.7 7 are required.
Lewis, Senior Regulatory Device & Biologics Expert — On October 20, 2023, FDA announced the availability of the final guidance authored by CBER titled “Voluntary Consensus Standards Recognition Program for Regenerative Medicine Therapies.” Existing published VCS may be identified internally by FDA or externally by stakeholders.
And, because it also manufactures products, its staff is current on regulatory requirements and FDA/ICH Guidance documents. Fully integrated development and manufacturing partners can minimise the impact of delays, improve overall efficiency, as well as minimising FDA information requests to enable a faster approval cycle.
Enbrel is approved by the Food and Drug Administration (FDA) to treat moderate to severe rheumatoid arthritis , psoriatic arthritis, ankylosing spondylitis, polyarticular juvenile idiopathic arthritis, and moderate to severe plaque psoriasis. Check with your airline, though, about the documentation needed.
The FDA, PIC/S, and WHO have all emphasized the importance and benefits of data flows in their guidance on data integrity. 3 , 4 RAMI integrates all assets, including physical items, software, administrative shell, documents, and personnel. In the fourth and last step, the defined work packages are executed. to Industry 4.0
Wegovy is a brand-name medication approved by the Food and Drug Administration (FDA) for weight loss in certain patients, including children 12 and older. per package, which is equivalent to about $16,188 per year. It should be used together with a reduced-calorie diet and increased physical activity. How much does Wegovy cost?
Concept Paper: M4Q(R2) Common Technical Document on Quality Guideline.” As part of the US Food and Drug Administration (FDA) QbD pilot 4 in 2005–2006, the need to convey how the control strategy is linked to the target product profile (TPP) and quality target product profile (QTPP) was discussed. The authors propose using Module 2.3
Concept Paper: M4Q(R2) Common Technical Document on Quality Guideline.” As part of the US Food and Drug Administration (FDA) QbD pilot 4 in 2005–2006, the need to convey how the control strategy is linked to the target product profile (TPP) and quality target product profile (QTPP) was discussed. The authors propose using Module 2.3
When the Food and Drug Administration (FDA) approved Mounjaro (tirzepatide) in 2022 to treat Type 2 diabetes, some people hailed the GIP and GLP-1 medication as the next great antidiabetic drug. Samples often come in a package of four 2.5 And it’s not just hype. mg single-use pens, a one-month supply.
Hosted software to publish regulatory submissions in the electronic common technical document (eCTD) format was deployed. The group successfully submitted a “pilot” eCTD submission to the Food and Drug Administration (FDA) and gained FDA approval to transmit submissions through the Electronic Submissions Gateway (ESG).
While both Qulipta and Nurtec have similar mechanisms of action and are approved by the Food and Drug Administration (FDA) for migraine treatment , they have distinct applications: Qulipta is mainly used for the prevention of episodic migraines, whereas Nurtec can both prevent and treat acute migraine episodes.
For example, the equipment for blending and packaging large-volume, high-viscosity formulations is complex, and the residues can be challenging to remove. Returning to a development process within the execution of a validation protocol can come with complexities in documentation and regulatory compliance.
The setpoint for “proper” air velocity in cleanroom systems is documented in standards and regulations as 0.45 9 In 2015, the FDA published a guidance manual 10 that provided questions for inspections, including: Is the air flow in critical areas unidirectional when delivered to the point of use? m/s up to 0.54 At what velocity?
The chosen CxP focused on finding solutions and resolving issues instead of only documenting problems, which proved beneficial in turning over fully functional and highly integrated facilities for critically complicated projects already within the company’s portfolio. To infinity and beyond, indeed.
For changes requiring approval before implementation, once the necessary information has been submitted across regions, each health authority must review the data package and documentation based on local requirements. laboratory notebooks, batch records, and technical reports) to submission documents (e.g.,
Why, then, are FDA inspections abroad so infrequent and disappointing? FDA Inspections Suspended: During the COVID pandemic, the Food and Drug Administration suspended its inspections at most foreign pharmaceutical facilities. Even under the best of conditions, the FDA never inspects all foreign pharmaceutical plants annually.
The medication is available in both brand-name and compounded forms, although the compounded forms are not FDA approved. Some insurance plans may cover Ozempic for managing Type 2 diabetes , which is an indication that is approved by the Food and Drug Administration (FDA) to treat. Does insurance cover Ozempic?
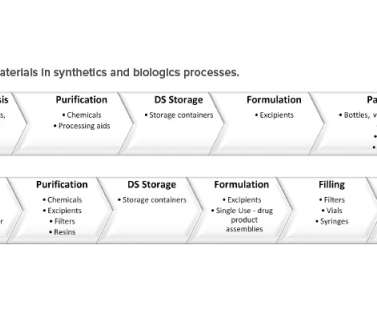
In this article, the term “raw material” refers to a material used in the manufacturing and packaging of a drug substance (DS) or a drug product (DP). Finally, the DP is packaged in a suitable container to ensure continued quality. It is considered a moderate change by the FDA (CBE30) and NMPA.
The past year has seen significant developments in the injectables landscape with the rapid introduction and development of vaccines in response to the pandemic, updates in regulations including the EU MDR and FDA guidance on bridging studies, and increasing industry acceptance of connectivity to aid the user experience. Secondary packagers.
In response to supply chain challenges, including shortages of critical glass vials for products like COVID-19 vaccines and parenteral drugs, the Packaging and Distribution Expert Committee (PD EC) has made a significant revision of United States Pharmacopeia (USP) General Chapter <660>—Glass.
“However, since children’s dosing recommendations may vary between Benadryl products, it’s best to check the package label for dosing instructions to ensure appropriate and safe dosing,” says Dr. Johnson-Arbor. Dosing for the topical versions of Benadryl will differ, so always follow package instructions or consult your healthcare provider.
“Antioxidant” has become a hyper-common wellness buzzword, slapped on health food packages so often that many people don’t think twice about it. 10 potential health benefits of NAC Some of NAC’s uses are time-tested and well-documented, like its efficacy in treating acetaminophen overdose.
Thankfully, paper-based studies are becoming a thing of the past, but spreadsheets and Word documents are still commonly used to design and build trials. These are now used by several regulatory bodies, including the Food and Drug Administration (FDA) and the Japanese Pharmaceuticals and Medical Devices Agency (PMDA) for submission packages.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.











































Let's personalize your content