This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The brief, 20-page document, instead, focuses on AI models used to produce data that supports regulatory decision-making about the safety, effectiveness, or quality of drugs. The wastewater continued to contain high concentrations of organofluorines and other compounds that meet the definition of PFAS even after treatment.
If you work in pharma, the chances are you’re no stranger to the United States Food and Drug Administration, or FDA, which regulates pharmaceuticals. While the FDA is responsible for regulating both drugs and devices, they’re handled through completely different processes in different parts of the agency.
Baumhardt, Senior Medical Device Regulation Expert — With the explosion of health‑related software, many software developers are generating products with functionality that is subject to regulation by the Food and Drug Administration (FDA). Please check out FDA’s presentation on this very topic – Is My Product a Medical Device?
Baumhardt, Senior Medical Device Regulation Expert — As an end of the year gift, FDA finalized its guidance document, Digital Health Technologies for Remote Data Acquisition in Clinical Investigations , in late December. The final guidance provides additional discussion with respect to DHTs that meet the definition of a device.
Food and Drug Administration (FDA) issued two guidance documents, one draft and one final, on food allergen labeling requirements. In addition to providing additional examples of tree nuts, the draft guidance states that FDA considers the following categories of fish to be major food allergens under Section 201(qq): Jawless fish (e.g.,
The primary vehicle for FDA to request cybersecurity information in premarket submissions has been guidance documents. The primary vehicle for FDA to request cybersecurity information in premarket submissions has been guidance documents. We have recently blogged on this topic (“ Is my software a medical device?
Food and Drug Administration (FDA) regulation as a medical device. Determining whether a product is a medical device subject to FDA regulation necessarily begins with understanding the FDA regulatory definition of ‘medical device’. a medical device subject to FDA oversight and enforcement.
Now, the FDA has published its first take on sourcing real-world data (RWD) from EHRs and medical claims, setting out its thinking on the approach that should be used to support regulatory filings for medicines. The FDA stresses it is a preliminary document and is encouraging comments, which can be filed up to 60 days after publication.
Lenz, Principal Medical Device Regulation Expert — FDA recently issued a draft guidance which would update the agency’s Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions guidance. FDA provides examples of changes that are likely and unlikely to impact cybersecurity of an existing device.
On 27th September 2022, the Food and Drug Administration (FDA) issued its final guidance for industry and FDA staff clinical decision support (CDS) software, which has been anticipated since the Center for Devices and Radiological Health (CDRH) listed the guidance as a top priority for fiscal year 2022. Criteria for regulation.
Mullen — More than five years after FDA first announced its plan to harmonize 21 CFR Part 820 with ISO 13485, on February 2, 2024, FDA finally issued the Quality Management System Regulation (QMSR) Final Rule. Both ISO 13485 and ISO 9000 contain terms and definitions that are referenced within Part 820. The new § 820.10
In 2006, FDA stated that DMCs were not recommended “for most clinical studies,” particularly those in early product development, short-term studies, or studies addressing less severe outcomes. In another update, the recent draft guidance added “entities reviewing safety data” and adaptation committees.
In the document, it was reasoned that global shortage of these vials threatened the delivery of COVID-19 vaccines and availability of existing parenteral products.
Shapiro — More than a decade ago, FDA began systematically to incorporate review of human factors (HF) design validation within 510(k) reviews. Now FDA has issued a draft guidance , Content of Human Factors Information in Medical Device Marketing Submissions (Dec. FDA did not require HF data as a basis for clearance of the predicate.
By Riëtte van Laack — On September 28, 2022, FDA announced the availability of the proposed rule for the implied nutrient content claim “healthy.” The term healthy, as an implied nutrient claim, was first defined by FDA in 1994. FDA also announced it would be re-evaluating the regulatory criteria for use of the “healthy” claim.
A True Copy is an exact copy of original documentation that preserves the same content, meaning and attributes of the original. It is an electronic copy maintained in an electronic document management system. filling in a paper-based batch record or analytical result). However, for higher risk data (i.e.,
PM Counsel John Claud to testify yesterday about FDA’s foreign inspection program. We’ve blogged previously on the troubles FDA has faced ramping up its foreign inspections program after the pandemic. FDA was invited to attend but did not.
Javitt — On September 28, 2022, the FDA issued the long anticipated final Clinical Decision Support Software Guidance (CDS Guidance), which replaces the revised draft guidance document from 2019. While the final guidance includes the same definition of “signal” as that used in the draft guidance—i.e., Karst & Steven J.
In the US, botanical dietary supplements can be sold without US Food and Drug Administration (FDA) approval, 1 prompting many vendors to promote the use of botanical products as dietary supplements , 2 rather than pursue a path of regulatory approval. 3 Is two too few?
In May of 2014 and February of 2019 , the FDA released final Guidance for Industry outlining the Agency’s policies and procedures regarding expedited development and review programs for new drugs and biologics intended to treat serious or life-threatening conditions. Image courtesy of qimono at pixabay.com.
While a DEA registration is not required to sell these products, by definition, any person selling these products is a “regulated person” and all sales of these products are considered “regulated transactions.” 21 U.S.C. § 21 U.S.C. §§ 802(38), (39)(B). 1310.06.
Walsh — The question of whether FDA has authority to take pictures during an inspection of a facility has bounced around the food and drug bar for many years. FDA’s view is stated in the agency’s Investigation Operations Manual, and it is one with which a number of practitioners disagree. By Ricardo Carvajal & Anne K.
Bob also initiated an agreement under which ISPE’s Guidance Documents are made available to PIC/S and WHO inspectors, which is still in place today. Most recently he assisted the regulatory authorities of Saudi Arabia, Russia, China, Jordan, and Philippines in the process to attain PIC/S membership.
The first keynote speaker was Michael Kopcha, PhD, RPh, Director of the Office of Pharmaceutical Quality (OPQ), Center for Drug Evaluation and Research (CDER), US FDA. Quality is a foundation for the development and delivery of pharmaceuticals: per Kopcha’s definition, quality means consistently meeting the expectations of the user.
At the federal level, the Center of Veterinary Medicine (CVM) of FDA regulates food for animals, including livestock and pets. A major component of AAFCO is its work on ingredient definitions, specifying what ingredients may be use in animal feed under what conditions. This document also describes an appeal procedure.
Sasinowski — This time last year, we wrote about a long-overlooked FDA statutory authority and wondered if this provision, known colloquially as the “single study plus confirmatory evidence” pathway, was having a moment (previous post here ). Valentine & Frank J.
Valentine — As we begin the final quarter of 2022 and the leaves here on the east coast begin to turn and fall, it seems the clock may be running out on FDA and the Center for Drug Evaluation and Research (CDER) to meet its goal of publishing a draft guidance on confirmatory evidence this year. Sasinowski & James E.
It was usual practice for TGA to prepare additional guidance/explanatory documents for industry and for TGA inspection staff to participate in training seminars to assist industry. Under this definition, a regulatory authority need not adopt in totality any decision reached by another regulatory authority, but they make their own decision.
Small Dispensers and the 6/12/2024 FDA Announcement Concerning DSCSA This week’s article on the recent DSCSA postponement comes from PRS Pharmacy Services. If the FDA or another regulator requests DSCSA transaction data, the DSCSA regulations give you up to 48 hours to respond. The original article can be found HERE.
Lewis, Senior Regulatory Device & Biologics Expert — On October 20, 2023, FDA announced the availability of the final guidance authored by CBER titled “Voluntary Consensus Standards Recognition Program for Regenerative Medicine Therapies.” Existing published VCS may be identified internally by FDA or externally by stakeholders.
Another rejected proposal was a definition of “vaccine” (vaccines are exempt from Medicaid rebates), which would have limited this term to a product that is administered prophylactically – i.e., to prevent rather than treat a disease. Remarkably, manufacturers may not dispute a CMS notification. 1396r-8(k)(3).
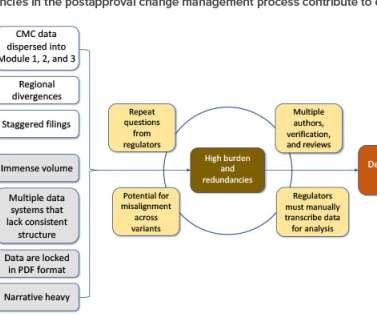
For changes requiring approval before implementation, once the necessary information has been submitted across regions, each health authority must review the data package and documentation based on local requirements. laboratory notebooks, batch records, and technical reports) to submission documents (e.g.,
The final presentation was Regulatory Considerations and Challenges in Development of Cell Therapy Products – CMC Perspective delivered by Dr. Melanie Eacho, Cell Therapies Branch Chief within the Office of Tissues and Advanced Therapies (OTAT) , US FDA/CBER. CDER/OPQ/OQS/FDA. Christian Woelbeling. Executive Industry Advisor.
Once the designation request has been submitted to the FDA, it is reviewed by the OOPD , which determines if all criteria for ODD approval have been met. Each component of the application must be addressed completely and provide detailed information. The review period is 90 days from time of receipt.
Concept Paper: M4Q(R2) Common Technical Document on Quality Guideline.” As part of the US Food and Drug Administration (FDA) QbD pilot 4 in 2005–2006, the need to convey how the control strategy is linked to the target product profile (TPP) and quality target product profile (QTPP) was discussed. The authors propose using Module 2.3
Concept Paper: M4Q(R2) Common Technical Document on Quality Guideline.” As part of the US Food and Drug Administration (FDA) QbD pilot 4 in 2005–2006, the need to convey how the control strategy is linked to the target product profile (TPP) and quality target product profile (QTPP) was discussed. The authors propose using Module 2.3
The Act also established new definitions for “Internet,” “online pharmacy,” “practice of telemedicine,” among others. Comment topics suggested by DEA include the following: Whether the rule should limit the issuance of prescriptions for controlled medications to FDA-approved indications contained in the labeling for those medications.
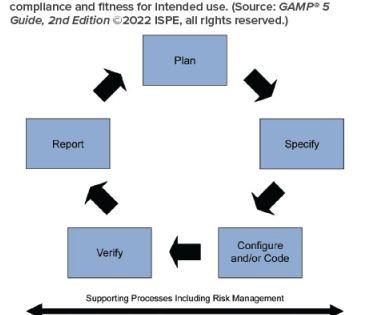
The ISPE Sub-Committee for Paperless Validation has defined ‘Paperless Validation Systems’ as Paperless solutions enable validation lifecycle deliverables to be generated, approved, and more importantly, testing to be completed without the need for the printing of paper test documents. If It Is Not Documented, It Did Not Happen.
Collection Device Special Controls Sponsor will need to select a collection device that is either FDA-cleared, -approved, or -classified as 510(k) exempt (either as a standalone collection device or as part of a system) or alternatively the collection device can be cleared within the premarket submission for the POC test.
For example, an assessment for a facility primarily focused on US and EU markets would only consider GMPs and regulations from EudraLex and the FDA. FDA CFR Title 21 Parts 211, 600, and 1271; 8. , FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing–Current Good Manufacturing Practice, 11.
Q13 Development Timeline The process of drafting the new guidance document was initiated in earnest in November 2018 when the concept paper 2 and business plan were endorsed. Since then, agencies have begun officially adopting the guidance, with the FDA doing so in March 2023.
Also, determining whether an intrusion occurred and documenting that intrusion adds to the challenges of performing aseptic technique properly (and perhaps adds to the subjectivity of the current control strategies). Currently, no definitive method for ML software validation has been established by either regulators or industry.
It was usual practice for TGA to prepare additional guidance/explanatory documents for industry and for TGA inspection staff to participate in training seminars to assist industry. Under this definition, a regulatory authority need not adopt in totality any decision reached by another regulatory authority, but they make their own decision.
It was time for ISPE to update this key guidance document to reflect technological progress. Concepts of computerized software assurance (CSA) as discussed in the US FDA Center for Devices and Radiological Health (CDRH) Case for Quality program 1 are also explored and applied. 1 US Food and Drug Administration. 15 September 2020.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.















































Let's personalize your content