This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Walsh — Among FDA-regulated establishments and stakeholders, there is one word that makes everyone go on edge – the dreaded FDA “inspection.” In FY2023, FDA conducted over 1000 inspections under the BIMO program. We also detail some of our recommended best practices to achieve success when FDA comes knocking.
Schwartz — FDA recently published a Federal Register (FR) Notice [ Docket No. FDA-2024-N-3945 ] announcing the publication of a draft strategy document, for public comment, outlining specific actions FDA plans to take to facilitate the use of innovative manufacturing technologies.
On February 7, at a town hall organised to discuss clinical trial designs for gene therapies, FDA experts pushed pharma players to look for ways to establish clinical effectiveness despite the challenges in recruiting patients with rare diseases.
After a firm submits a 510(k) to FDA, FDA will request still more information after a first-pass review. According to the 2 nd Quarter FY2023 MDUFA V Performance Report , FDA issued a request for additional information (AI request) on the first FDA review cycle for 63% to 68% of 510(k)s submitted in FY2018 to FY2022.
Baumhardt, Senior Medical Device Regulation Expert — With the explosion of health‑related software, many software developers are generating products with functionality that is subject to regulation by the Food and Drug Administration (FDA). Please check out FDA’s presentation on this very topic – Is My Product a Medical Device?
In 2006, FDA stated that DMCs were not recommended “for most clinical studies,” particularly those in early product development, short-term studies, or studies addressing less severe outcomes. In another update, the recent draft guidance added “entities reviewing safety data” and adaptation committees.
The primary vehicle for FDA to request cybersecurity information in premarket submissions has been guidance documents. The primary vehicle for FDA to request cybersecurity information in premarket submissions has been guidance documents. It applies whether the software is the entire device (i.e.,
Walsh — Last fall, we blogged about the process FDA uses to review allegations of regulatory misconduct against device manufacturers, suggesting greater transparency on the FDA process was needed (see here ).
It appears that the FDA is inclined to support Biogen’s amyotrophic lateral sclerosis (ALS) drug, tofersen, with documents released on Monday stressing the need for “regulatory flexibility” when assessing treatments for life threatening diseases such as ALS. read more
Mullen — On June 2, 2023, FDA issued the latest version of its guidance on Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program. The document provides minor updates on procedures, incorporating recent developments and experiences in how FDA interacts with industry.
On 27th September 2022, the Food and Drug Administration (FDA) issued its final guidance for industry and FDA staff clinical decision support (CDS) software, which has been anticipated since the Center for Devices and Radiological Health (CDRH) listed the guidance as a top priority for fiscal year 2022. Criteria for regulation.
Continue active cooperation with strategic partners , encourage PIC/S membership and strengthen PIC/S communications and engagement. Build PIC/S operational capacity by securing new revenue streams, implementing fiscally sound financial plans and increase transparency to the PIC/S community.
Lenz, Principal Medical Device Regulation Expert — FDA began the Software Precertification (Pre-Cert) pilot program in 2017 to evaluate an alternative approach to regulation of software as a medical device (SaMD) over the total product lifecycle (TPLC). As part of the pilot program, FDA included a Test Plan. By Adrienne R.
In May of 2014 and February of 2019 , the FDA released final Guidance for Industry outlining the Agency’s policies and procedures regarding expedited development and review programs for new drugs and biologics intended to treat serious or life-threatening conditions. Image courtesy of qimono at pixabay.com.
If multiple people are reviewing and need to communicate changes it may be challenging to ensure all updates are incorporated. Lack of compatibility with cloud-based document sharing. Creation of unnecessary documents. There is no option to reference a document that has already been attached elsewhere in the submission.
Javitt — On September 28, 2022, the FDA issued the long anticipated final Clinical Decision Support Software Guidance (CDS Guidance), which replaces the revised draft guidance document from 2019. FDA interprets the term “pattern” to mean “multiple, sequential, or repeated measurements of a signal or from a signal acquisition system.”
The pandemic had changed the way people live, work and communicate. FDA has begun to reintroduce in-person meetings. It is also noteworthy that in the March 27 announcement , FDA distinguished between teleconferences and videoconferences despite using the same online platform (e.g., However, not all changes are forever.
The US Food and Drug Administration (FDA) has released the fourth and final chapter in a series of guidance documents designed to support patient-focused drug development. The guidance also touched upon clinical trial design elements that must be explained, described, or rationalised to the FDA.
User fees are fees that FDA collects from “companies that produce certain products, such as drugs and medical devices, and from some other entities, such as certain accreditation and certification bodies.” These fees supplement the annual funding Congress provides for FDA. These fees are negotiated by FDA and industry every five years.
There are also numerous industry guidance documents for products manufactured for use in clinical trials and guidelines, some which are specific to advanced therapeutic drug products that must be consulted. Teams must also accept the variety of analytical procedures these regulatory agencies prefer.
These types of pharmacies act as the contact between the pharmaceutical company and regulatory agencies, such as the FDA. Assessing and communicating the regulatory guidelines for medication approval. Presenting documents to the appropriate regulatory agency and making changes as necessary .
That’s because the Food and Drug Administration (FDA) has approved it for Type 2 diabetes , while weight management is technically an off-label treatment. Foxman recommends that every patient review their formulary to see how Ozempic is covered and communicate directly with their insurer for clarity on the specific requirements. “It’s
Lenz, Principal Medical Device Regulation Expert — Since 2005, 510(k) submissions have been formatted according to FDA guidance Format for Traditional and Abbreviated 510(k)s. Several sections of the eSTAR templates have questions that walk through related guidance documents. By Adrienne R. Know Your Guidances.
Her expertise includes preparation and maintenance of licensing documentation, communications with state boards and government agencies, conducting research, and the preparation of associated licensing documentation and applications.
The document provides specific guidance on the sampling and testing procedures for environmental monitoring, as well as the limits for particular and microbial contamination. Some of these challenges include for example regulatory compliance, quality control and effective communication between the original manufacturer and the CMO.
By Philip Won & Véronique Li, Senior Medical Device Regulation Expert — As we recently blogged , FDA released three draft guidance documents to help enhance the predictability, consistency, and transparency of the 510(k) program. One of these documents focuses on “ Evidentiary Expectations for 510(k) Implant Devices.”
Medical affairs in Pharma are often seen as a central agency that works within a healthcare company and prioritize communication among life science organizations, medical professionals, healthcare providers, and patients. Medical affairs definition uses clinical and scientific information to communicate the efficiency of a drug.
Small Dispensers and the 6/12/2024 FDA Announcement Concerning DSCSA This week’s article on the recent DSCSA postponement comes from PRS Pharmacy Services. If the FDA or another regulator requests DSCSA transaction data, the DSCSA regulations give you up to 48 hours to respond. The original article can be found HERE.
According to the legal documents , the plaintiffs accused the companies of “violating federal antitrust laws, alleging “per se” and “rule of reason” violations”. Gilenya is an oral medication for multiple sclerosis. Fighting Entresto generics. Please check your email to download the Report.
Two licensed pharmacists must directly communicate the transfer. As with other required controlled substance records, electronic records documenting EPCS transfers must be maintained for two years from the transfer date by both the transferring and receiving pharmacies. cannot be altered during the transmission.
Lewis, Senior Regulatory Device & Biologics Expert — On October 20, 2023, FDA announced the availability of the final guidance authored by CBER titled “Voluntary Consensus Standards Recognition Program for Regenerative Medicine Therapies.” Existing published VCS may be identified internally by FDA or externally by stakeholders.
Regulatory bodies such as the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) place great importance on data integrity in the pharmaceutical/life science industry. Good documentation practices. Joseph S Boakai (JSB): The three current trends are as follows: Increased scrutiny by regulatory bodies.
The Advancing Pharmaceutical Quality, Quality Management Maturity Program includes five guidance documents: Corrective Action and Preventive Action (CAPA) : ICH Q10 demonstrates defined requirements for a robust corrective action and preventive action system throughout the product lifecycle. Internal Communication. CAPA Effectiveness.
The standard practice is for FDA to engage with companies on an individual basis through formal meetings and deliver directed feedback to specific sponsor questions. OTP has spent years communicating industry-wide problems on an individual basis. At this time, they recommend developers follow ICH Q5e when assessing comparability.
Yes, as of March 2020 , insulin is considered a biologic product by the FDA. Regulatory bodies such as the FDA conduct thorough tests to confirm that they meet safety, purity, and potency standards. The FDA Purple Book database can be a good resource to determine whether a biologic drug is interchangeable.
Achieving effective communication between a healthcare specialist and a patient is impossible if data the healthcare provider relies on data that can be misunderstood, misinterpreted, and misused. If any piece of data is used after its documentation or creation, it’s crucial that it be attributed to the original source.
Concept Paper: M4Q(R2) Common Technical Document on Quality Guideline.” As part of the US Food and Drug Administration (FDA) QbD pilot 4 in 2005–2006, the need to convey how the control strategy is linked to the target product profile (TPP) and quality target product profile (QTPP) was discussed. The authors propose using Module 2.3
Concept Paper: M4Q(R2) Common Technical Document on Quality Guideline.” As part of the US Food and Drug Administration (FDA) QbD pilot 4 in 2005–2006, the need to convey how the control strategy is linked to the target product profile (TPP) and quality target product profile (QTPP) was discussed. The authors propose using Module 2.3
They also used direct communication, marketing materials, media and financial statements, models and other information to defraud potential investors, according to the complaint. She admitted using pharma company logos – including Pfizer – on Theranos documents sent to investors, even though they had not endorsed the technology.
This record-keeping includes detailed documentation of the product’s history and movement through the supply chain. When regulators, such as the FDA, request information about a specific drug product, your pharmacy must be able to promptly provide the necessary traceability data. The DSCSA aims to streamline this recall process.
The FDA has approved these devices for mild to moderate hearing loss. Necessary documentation and information Financial assistance programs may require proof of income. Other documentation they may require includes: Identification (driver’s license, passport, etc.)
Improvements have been observed by various regulatory agencies in Latin America for the acceptance and implementation of international standards—for instance, the ICH Common Technical Document (CTD) format. Additional required information beyond ICH guidelines, or non-value-added documents (e.g., 6, and 2.3.S.7 7 are required.
A lack of transparency The importance of setting the primary outcome prior to commencing a study and not deviating from the original protocol, was first highlighted in 1990 by Jay Siegel , a physician and research scientist working for the FDA in the US. Nevertheless, it is incumbent on trialists to communicate such changes.
and management communication strategy. This includes ensuring maturity levels for all operating model elements follow the five general maturity levels defined by the FDA: 1–Initial, 2–Developmental, 3–Defined, 4–Managed, and 5–Optimized. Is the vendors’ documentation complete in order to assess GxP risks and GxP compliance?
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.























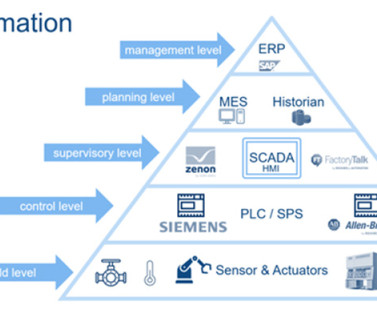
























Let's personalize your content