This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Food and Drug Administration (FDA) issued two guidance documents, one draft and one final, on food allergen labeling requirements. 1, 2023; The applicability of food allergen labeling requirements to specific products (e.g., 1, 2023; The applicability of food allergen labeling requirements to specific products (e.g.,
Koblitz — Well, we’re a little late to blogging about this, but the significance of the ongoing Teva v. GSK litigation to Hatch-Waxman aficionados makes this case still ripe for blogging. GSK skinny label case , the U.S. Six months after the Supreme Court asked the Solicitor General to submit a brief on behalf of the U.S.
Memorizing 45+ page document is certainly not a reasonable expectation, but one can certainly walk away with an awareness of general concepts and themes which are relevant. ” Labeling the use of an antibiotic as inappropriate or appropriate cannot simply be done based upon whether it is FDA-approved for a given indication.
Shapiro — Last summer, FDA published a draft guidance, Laser-Assisted In Situ Keratomileusis (LASIK) Lasers – Patient Labeling Recommendations (July 29, 2022) setting forth a proposal for new recommended patient‑directed labeling. The approved devices already have patient‑directed labeling that FDA has approved. By Jeffrey K.
The primary vehicle for FDA to request cybersecurity information in premarket submissions has been guidance documents. We have recently blogged on this topic (“ Is my software a medical device? ”). Per section 3305(e) of the Omnibus, FDA must provide an updated guidance document by December 2024. Loose Ends IDEs.
By Véronique Li, Senior Medical Device Regulation Expert & Philip Won — Last week, our blog post advised planning a transition strategy in advance of the news of the termination of the Covid-19 public health emergency. These are documents on the A-list, a list of prioritized documents that FDA intends to publish during FY2023.
Part 820), labeling (21 C.F.R. In practice, a standard operating procedure for design controls is developed to describe how medical device design will be performed and documented. First, a design history file includes many documents that must be submitted as part of a 510(k), De Novo, or premarket approval (PMA) application.
Walsh — Last fall, we blogged about the process FDA uses to review allegations of regulatory misconduct against device manufacturers, suggesting greater transparency on the FDA process was needed (see here ). FDA also requests a “detailed description of the allegation with any available supporting documentation.”
The long-awaited final rule, which we last discussed in a July 2023 blog post and have tracked in our March 2023 and March 2022 posts, aims to harmonize quality management system requirements for medical devices with requirements set forth by other regulatory authorities around the world. Notably, Part 820 will look different. The new § 820.10
Section IV, Additional Resources, provides links to previously-issued guidance documents and other educational materials geared to traditional device manufacturers, with no additional commentary on how to apply these requirements to the very different clinical laboratory environment.
Blogs – A company is free to own a blog or engage a blog writer by way of sponsorship or consultancy fees. In the EU and UK, where direct-to-consumer promotion of POMs is prohibited, companies may not sponsor blogs that promote, or could reasonably be expected to promote, such products.
The general structure of our submissions, and of many others that we have seen, is to have a document for each of the twenty sections and then to refer to other files, such as labeling and test reports, as attachments. Several sections of the eSTAR templates have questions that walk through related guidance documents.
In this blog post, we will first briefly outline the procedural steps in the 510(k) review process for medical devices. However, the strategy of addressing the AI request presented in the next section of this blog post applies to all three types of 510(k)s. FDA Day (in calendar days) FDA Actions Day 1 FDA receives 510(k) submission.
Examples of where we would generally consider preventing the FDA investigator from fully conducting an inspection include… [t]he owner, operator, or agent in charge refuses to allow the FDA investigator to collect evidence to document potential violations (e.g., That’s a long time to potentially lose access to the U.S. See here and here.
a) , and related guidance documents (e.g., Post-market, manufacturers can make modifications consistent with the PCCP and document the modification in accordance with their quality system, without the need for a new marketing submission. See 21 CFR 807.81(a)(3) a)(3) and 21 CFR 814.39(a)
The new draft guidance continues to borrow heavily from the NDA process and FDA notes that it used the same source material on which other drug application recommendations are based including the Common Technical Document (CTD). x 11-inch paper, and with hyperlinks to references and numbered pages.
Javitt — On September 28, 2022, the FDA issued the long anticipated final Clinical Decision Support Software Guidance (CDS Guidance), which replaces the revised draft guidance document from 2019. You can find our preliminary blog post on the release of this guidance here , and our blog posts on the draft CDS guidances here and here.
By Philip Won & Véronique Li, Senior Medical Device Regulation Expert — As we recently blogged , FDA released three draft guidance documents to help enhance the predictability, consistency, and transparency of the 510(k) program. One of these documents focuses on “ Evidentiary Expectations for 510(k) Implant Devices.”
Such codes need to be placed on device labels and packages to allow devices to be easily identified and tracked throughout their lifecycle, except where the rule provided for an exception or alternative. The compliance dates were first published in 2013, and subsequently updated in various guidance documents and regulations published by FDA.
It also presents specific questions to collect data from the submitter and provides links to relevant regulations and guidance documents. To learn more about our experience with the eSTAR for 510(k)s, check out our previous blogs ( here , here ).
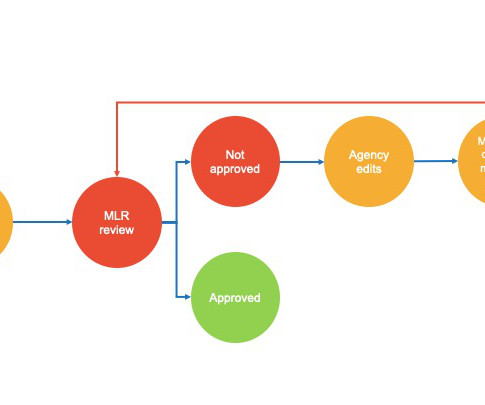
billion for the illegal marketing of four of its off-label drugs, which became one of the biggest fraud settlements in the healthcare industry. The truth is that MLR reviews are needed in 90% of cases, whether it is a website blog post or an important research paper. For example, Pfizer was fined $2.3
• Auto Bar-Code Labeling produced only for final preparations. • Auto Documentation timestamps each preparation step serving as a black box and assists in fulfilling USP documentation requirements. . • Volume Verification that ensure base solutions and additives are correct, (e.g.,
For one thing, anesthesia workstations include safety features, such as the ability to print out color-coded labels for syringes, which can help prevent harmful medication errors. We’ve developed closed-loop functionality that makes it easier for providers to document waste, improving the likelihood of compliance.
States frequently review labels (and labeling) for animal food products. The CFI includes a list of common foods that “may be appropriate for use in animal food and serve as a tool for use during review of ingredients on an animal food label.” This document also describes an appeal procedure.
In 2015, FDA issued a Warning Letter to Kind LLC because, among other things, the company labeled products as healthy whereas these products provided more saturated fat per serving than permitted by the regulatory definition. It is focused on the nutrients in the food product rather than on it overall nutritional “quality.”.
Omnicell continues to collaborate with ISMP to improve pharmacy-nursing efficiency, reduce medication errors during dispensing, and support accurate medication tracking and documentation of inventory and deter drug diversion. Watch this webinar to learn more about how automated dispensing systems support ISMP guidelines for medication safety.
Nor does FDA have the authority to look beyond the Lumryz labeling to its REMS document for a use that overlaps with the listed use code, Avadel argues. Another interesting question this suit raises is whether FDA can look beyond the labeling to determine whether a section viii carve-out is acceptable.
In fact, the priority designation for the final rule is labeled as “economically significant.” Finally, CDRH would need to ensure alignment on existing guidance documents and regulations that refer to the QSR or 21 C.F.R. Dr. Shuren further elaborated that he hopes the final rule will be “out by the end of this year.”
Even in the absence of a guidance document dedicated to explaining FDA’s views on interpreting the statutory construct of “confirmatory evidence,” a number of drug approvals over the past few years give us a great deal of insight into FDA’s current thinking and how FDA is applying this standard.
On July 7, 2023, armed with these new procedures, CDER notified Oncopeptides that it proposed expedited withdrawal of Pepaxto because the postapproval study failed to verify clinical benefit and because Pepaxto was not shown to be safe or effective under its conditions of use (the documents discussed herein are published in the docket here ).
In this blog we examine the Special Controls put in place to mitigate false results, incorrect interpretation of results, and incorrect operation of the device. The monitoring protocol also needs to include plans to update labeling with the additional performance data. Customer Support Help line).
The new guidance is one of three policy documents dedicated to explaining FDA’s interpretation of this statutory authority and their approach to exercising scientific judgment in evaluating drug effectiveness. We are heartened to see that this latest guidance reflects many of the advances we observed in practice since 2019.
The only exception for a modified device is when there were no changes to: the user interface; intended device users; intended device uses; intended use environment(s); training or labeling. as a de novo special control or in a guidance document for a specific device type).
Two device manufacturers received Warning Letters for QSR violations (QSR is FDA’s label for cGMP requirements applicable to medical device manufacturers). market complaints in 2021 through 2023 but could not provide documentation to demonstrate how the complaints were reviewed or evaluated. Terragene S.A., Terragene S.A.,
Food and Drug Administration – labeled an “Official Action Indicated” classification – is generally devastating for the facility, not least because it can stall FDA approval of applications to market drugs manufactured at the facility. By Douglas B. Farquhar — A drug manufacturer’s bad post-inspection grade from the U.S.
The Draft Guidance continues to list the regulatory requirements to obtain authorization for charging in a clinical trial setting: (1) evidence that the drug has a potential clinical benefit that would provide a significant advantage over available products; (2) the data to be obtained from the trial would be essential to establishing that the drug (..)
CBER may still request additional information when deemed appropriate, but the stated hope is that increased use of VCS can facilitate product development by reducing the need to develop unique methods for individual products and that they will typically reduce the amount of necessary documentation “and may reduce FDA review time.”
Acute cardiac injury has been a well-documented feature of infection with SARS-CoV-2, which causes COVID-19 among patients requiring hospitalization. Back to Main Blog. One area of concern for both health care providers and their patients is the long-term effect of COVID-19 infection on patients with chronic diseases. About the author.
These documents focused on why the sentinel event occurred. Both the label and the product had to match the order before the product. We found nothing wrong with the IV solutions or their labeling. But when everything becomes a sentinel event, it sort of loses something in its effectiveness.
Comment topics suggested by DEA include the following: Whether the rule should limit the issuance of prescriptions for controlled medications to FDA-approved indications contained in the labeling for those medications. Helpful practitioner prescribing guidance is set forth in a simplified document here. Stay tuned.
The Board is not required to identify all drugs that meet the above criteria and may, in consultation with the advisory council and the commissioner of health, identify drug products that fall outside these criteria but otherwise could create “significant affordability challenges” for Minnesota’s healthcare system or Minnesota patients.
I was inspired to write this blog after the September 2020 Cameron Peak fires in Colorado. Perhaps more concerning, of the 133 different VOCs found across all tested products, only one, ethanol, was actually listed on any label! (13, For sunscreens and other over-the-counter drugs, fragrances must be identified on the label.
So, it ensures that you always have a steady stream of content posted as evergreen content, such as blog posts, podcasts, and videos. . Labeling systems. If your company’s content calendar has a rainbow of colorful labels, translate your organizational system to your scheduling tool. Hashtag groups. Key Features.
So, it ensures that you always have a steady stream of content posted as evergreen content, such as blog posts, podcasts, and videos. . Labeling systems. If your company’s content calendar has a rainbow of colorful labels, translate your organizational system to your scheduling tool. Hashtag groups. Key Features.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.

















































Let's personalize your content