This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
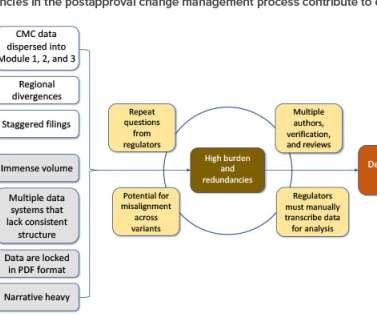
Following approval of an initial marketing application, postapproval changes are needed to ensure adequate supply, mitigate supply risk, expand patient market access, optimize manufacturing processes, improve analytical methods, and comply with new regulatory expectations. Common Technical Document [CTD] sections).
Concept Paper: M4Q(R2) Common Technical Document on Quality Guideline.” This article summarizes an alternate and more functional way to format the QOS presented in Module 2.3. The authors propose using Module 2.3 3 1 International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use.
Concept Paper: M4Q(R2) Common Technical Document on Quality Guideline.” This article summarizes an alternate and more functional way to format the QOS presented in Module 2.3. The authors propose using Module 2.3 3 1 International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use.
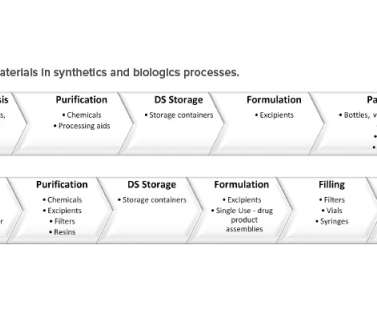
In this article, the term “raw material” refers to a material used in the manufacturing and packaging of a drug substance (DS) or a drug product (DP). This article highlights the regulatory expectations of raw materials, the challenges of postapproval changes. and the impact on supply resiliency.
This article presents relevant insights on the current regulatory and technical landscape for decentralized manufacturing, with select examples of current applications, and discusses perspectives on evolving and adapting the current regulations to meet future capabilities. Product Focus: Advancing Aseptic Processing.” 5 October 2022.
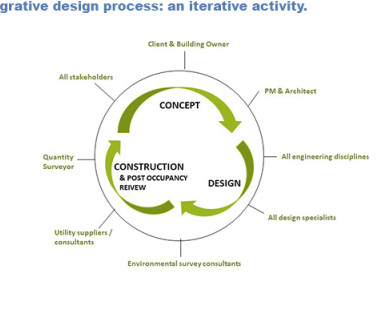
linear) design process is central to successfully attaining such comprehensive sustainability goals and project needs (see Figure 1). Process Intensification in the Biopharma Industry: Improving Efficiency of Protein Manufacturing Processes from Development to Production Scale Using Synergistic Approaches.”
These long global approval timelines complicate supply chain management by delaying innovations that improve quality assurance and by increasing the potential for supply interruptions and shortages that impact patient access to products. The proposals are consistent with WHO GRP. Published March 2020. www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-q12-technical-regulatory-considerations-pharmaceutical-product-lifecycle-management_en.pdf
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.











Let's personalize your content