This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
In this post a top 10 list of journal articles to read during an infectious diseases pharmacy rotation is provided. Articles included here are some of my favorites. Not being included in this list does not reflect a lack of quality and certainly there are many robust articles that could be added. Authored by: Timothy P.
Gonzalez — The Wall Street Journal (WSJ) recently published a series of articles as part of its special report “What’s Ahead for Artificial Intelligence.” Three of these articles focus on medical applications of Artificial Intelligence and Machine Learning (AI/ML) and explore FDA’s role in regulating such products.
The US Food and Drug Administration (FDA) created live biotherapeutic products (LBP) as a new category in the 2012 guidelines. EMA and FDA have given prime importance to the whole genome sequence characterisation of the strain in the final product dossier. Download and read more now… .
Baumhardt, Senior Medical Device Regulation Expert — With the explosion of health‑related software, many software developers are generating products with functionality that is subject to regulation by the Food and Drug Administration (FDA). Please check out FDA’s presentation on this very topic – Is My Product a Medical Device?
Food and Drug Administration (FDA) regulation as a medical device. Determining whether a product is a medical device subject to FDA regulation necessarily begins with understanding the FDA regulatory definition of ‘medical device’. When assessing mHealth technology, the FDA will determine whether an application is either: i.
Why, then, are FDA inspections abroad so infrequent and disappointing? FDA Inspections Suspended: During the COVID pandemic, the Food and Drug Administration suspended its inspections at most foreign pharmaceutical facilities. Even under the best of conditions, the FDA never inspects all foreign pharmaceutical plants annually.
The medication is available in both brand-name and compounded forms, although the compounded forms are not FDA approved. In this article, well take a closer look at semaglutide, its costs, and alternatives that may be offered. Learn more from our article on how to get Wegovy covered by insurance. Does insurance cover Ozempic?
The assessment report of the Committee for Medicinal Products for Human Use (CHMP)’s Article 5(3) of Regulation (EC) No 726/2004 opinion on nitrosamine impurities in human medicinal products offers guidance and recommendations on mitigation and prevention of nitrosamine-contaminated human medicinal products.
New research published in JAMA Health Forum has documented that the US National Institutes of Health (NIH) spent $8.1 billion for phased clinical trials of US Food and Drug Administration (FDA)-approved drugs between 2010-2019. This was ~10 percent of reported industry spending.
Validation projects have often been documentation-focused exercises, more befitting a grammar exercise than artifacts supportive of regulated activity. This article primarily discusses Appendix D5 Testing of Computerized Systems and Appendix M12 Critical Thinking. The GAMP 5 Second Edition includes: Six completely new appendices.
This article explores just one aspect of the RMP, that of RMMs, and how their effectiveness is measured with Dr Sophie Jouaville, an associate principal at IQVIA working on the design and oversight of non-interventional real-world evidence (RWE) safety and health economics and outcomes research (HEOR) studies.
Food and Drug Administration ( FDA ) as a weight loss medication. As of now, phentermine is not FDA-approved for ADHD, and the evidence of its effectiveness is scarce. Phentermine is an old psychotropic medication that is indicated by the FDA for the short-term treatment of weight loss,” she says. “It
Bob also initiated an agreement under which ISPE’s Guidance Documents are made available to PIC/S and WHO inspectors, which is still in place today. Most recently he assisted the regulatory authorities of Saudi Arabia, Russia, China, Jordan, and Philippines in the process to attain PIC/S membership.
Latuda (lurasidone) is a brand-name prescription drug that’s approved by the Food and Drug Administration (FDA) to treat schizophrenia as well as bipolar depression associated with bipolar I disorder. As of the time this article was written, Latuda’s average price for 30, 40 milligram (mg) tablets is $1,859. How much does Latuda cost?
This article will explore the advantages of using HCs, as well as several considerations for their effective use, including scientific methodology and regulatory guidance. FDA presentation. FDA Rare Diseases: Natural History Studies for Drug Development Guidance for Industry. Benefits of historical controls. Thompson, LA.
The past year has seen significant developments in the injectables landscape with the rapid introduction and development of vaccines in response to the pandemic, updates in regulations including the EU MDR and FDA guidance on bridging studies, and increasing industry acceptance of connectivity to aid the user experience. 08.30 – 12.30.
Small Dispensers and the 6/12/2024 FDA Announcement Concerning DSCSA This week’s article on the recent DSCSA postponement comes from PRS Pharmacy Services. The original article can be found HERE. If the FDA or another regulator requests DSCSA transaction data, the DSCSA regulations give you up to 48 hours to respond.
A lack of transparency The importance of setting the primary outcome prior to commencing a study and not deviating from the original protocol, was first highlighted in 1990 by Jay Siegel , a physician and research scientist working for the FDA in the US. Later work simply re-affirmed this finding.
We might conduct numerous interviews, look at thousands of documents, and travel to whatever locations are necessary. We prepare a report that we share with our client and make a recommendation to our client about whether to disclose the information to FDA. And, FDA still doesn’t.
The first keynote speaker was Michael Kopcha, PhD, RPh, Director of the Office of Pharmaceutical Quality (OPQ), Center for Drug Evaluation and Research (CDER), US FDA. The US FDA monitors quality, and Kopcha traced changes in regulation of quality over the years. FDA Activities in Support of Quality. Tue, 11/01/2022 - 06:49.
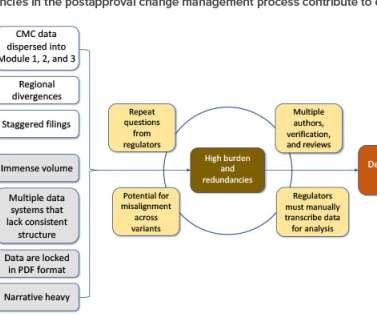
For changes requiring approval before implementation, once the necessary information has been submitted across regions, each health authority must review the data package and documentation based on local requirements. laboratory notebooks, batch records, and technical reports) to submission documents (e.g.,
This article will discuss pharmaceutical validation, its benefits, types, and basic recommendations. It also requires documenting the entire process, from raw material sourcing to product launch. It ensures compliance The Food and Drug Administration (FDA) in the U.S. Keep on reading!
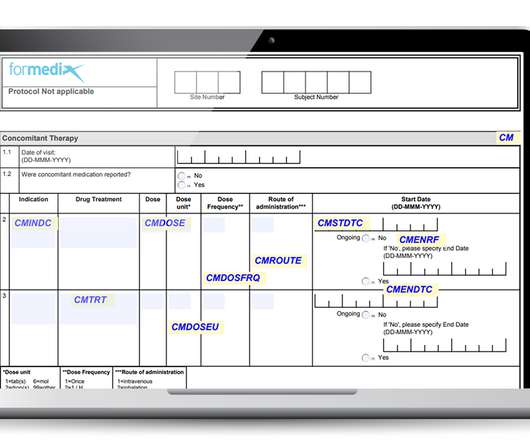
They must also comply with regulatory requirements, such as those defined by the FDA. Annotated CRFs are a key submission deliverable, a mandatory requirement of the FDA. In other words, “ each CRF should provide the variable names and coding for each CRF item included in the data tabulation datasets ” as stated by the FDA guidelines.
They must also comply with regulatory requirements, such as those defined by the FDA. Annotated CRFs are a key submission deliverable, a mandatory requirement of the FDA. In other words, “ each CRF should provide the variable names and coding for each CRF item included in the data tabulation datasets ” as stated by the FDA guidelines.
This error has appeared not just in articles and other news publications but in some court documents as well. In many cases, it appears DEA does not have the resources or infrastructure to maintain and produce the information to respond to FOIA requests as do other administrative agencies such as FDA.
The regulatory keynote was delivered by Peter Marks, MD, PhD, Director, Center for Biologics Evaluation and Research (CBER), US FDA. Marks outlined some concepts that the FDA is thinking about that could help move in this direction, such as developing a “cookbook” to standardize bespoke product development and manufacturing.
The panellists included: Roberto Conocchia GMP Technical Lead European Medicines Agency (EMA) Rick Friedman Deputy Director, Office of Manufacturing Quality FDA/CDER Alan Moon Lead Senior GMDP Inspector MHRA The discussions were moderated by Mark Birse and Jean-Francois Duliere. This is a key concern for the FDA.
This article outlines a broad framework to evaluate different types of modalities that may be accommodated concurrently in a new or existing facility and then uses a case study to explain how the approach may be applied to an existing facility. For the purposes of brevity, this article doesn’t discuss safety.
Improvements have been observed by various regulatory agencies in Latin America for the acceptance and implementation of international standards—for instance, the ICH Common Technical Document (CTD) format. This article offers an overview of the CMC requirements for the small molecules product registration process in Latin America.
This article is the first part of three and covers the first three of nine predictions, here focusing on global healthcare environmental challenges and what they mean for the pharmaceutical industry. China still hasn’t approved western mRNA vaccines, despite submission, and the US FDA rejects Chinese-only trial data for cancer medicines.
In our new article, you will learn more about data integrity in the pharmaceutical and life sciences industry and why maintaining it matters so much. Key Principles of Data Integrity In the 1990s, the FDA developed a list of principles of data integrity, which still serve as a framework for working with data and documenting it.
Concept Paper: M4Q(R2) Common Technical Document on Quality Guideline.” As part of the US Food and Drug Administration (FDA) QbD pilot 4 in 2005–2006, the need to convey how the control strategy is linked to the target product profile (TPP) and quality target product profile (QTPP) was discussed. The authors propose using Module 2.3
Concept Paper: M4Q(R2) Common Technical Document on Quality Guideline.” As part of the US Food and Drug Administration (FDA) QbD pilot 4 in 2005–2006, the need to convey how the control strategy is linked to the target product profile (TPP) and quality target product profile (QTPP) was discussed. The authors propose using Module 2.3
Food and Drug Administration (FDA) certifies the Red Dye 40 as safe for human consumption. Apart from Red Dye 40, there are eight other certified color additives approved for use in food by the FDA: Blue No. The FDA has denied any link between the two for years. The controversy surrounding Red Dye 40 The U.S. 1, Blue No.
Attendees were invited to submit questions to the FDA representatives. This article offers highlights from the discussion. Please note that views expressed by the panelists are not necessarily representative of the position of the FDA, and that questions and responses are lightly edited for clarity.
If you think that doesn’t happen nowadays, here’s an article about recent counterfeit HIV medications. This record-keeping includes detailed documentation of the product’s history and movement through the supply chain. Document Everything Meticulous record-keeping is a fundamental aspect of DSCSA compliance.
The US Food and Drug Administration (FDA) requires that manufacturers establish whether nitrosamines, including NDSRIs, could be present in active pharmaceutical ingredients (APIs) and drug products, using the “three-step mitigation strategy described in the agency’s guidance”. Nitrosamine Imp -Q&A-Mar-22.pdf.
Food and Drug Administration ( FDA ) for the treatment of schizophrenia , bipolar I disorder , autism spectrum disorders, Tourette syndrome, and as an add-on treatment for depression. This article provides six aripiprazole savings tips to help you afford your medication without breaking a sweat.
ISPE continues to build on its mission and commitment to providing content for the industry through guidance documents and its CoPs, including expanding CoP topics. Five successful conferences drove financial performance more than expected, and guidance documents have been very strong. Sherwood Article of the Year Award.
In this article, you will learn about: How LLLT works Existing LLLT research Some uses and benefits of LLLT for thyroid health Laser therapy in clinical settings and LED devices for at-home use Can Thyroid Tissue Really Regenerate? I want to share some information about LLLT with you and discuss some interesting research.
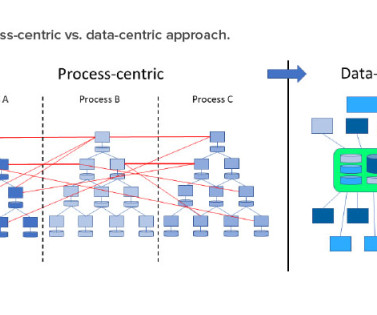
The FDA, PIC/S, and WHO have all emphasized the importance and benefits of data flows in their guidance on data integrity. This article provides insight into process maps and data flows in the biopharma industry using the Reference Architecture Model Industry 4.0 The concepts introduced here will be topics for future articles.
FDA/CBER/OMPT/OCBQ. And I'd like to make industry aware of a PIC/S document that's intended to guide inspectors on good data governance practices. Director, Office of Pharmaceutical Quality Operations. 2022 PIC/S Chair, Sr. Corporate Regulatory Compliance & Enforcement Advisor. Rick Friedman. Brooke Higgins. Robert Sausville.
This article highlights the overall benefits and scope of Q13, as well as what we see as the next set of opportunities to further expand the adoption of CM across the globe. Since then, agencies have begun officially adopting the guidance, with the FDA doing so in March 2023.
It was usual practice for TGA to prepare additional guidance/explanatory documents for industry and for TGA inspection staff to participate in training seminars to assist industry. The outcomes of these inspections were shared extensively with MRA partners such as EMA and US FDA. The approach by TGA and HSA were very similar.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.















































Let's personalize your content