This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
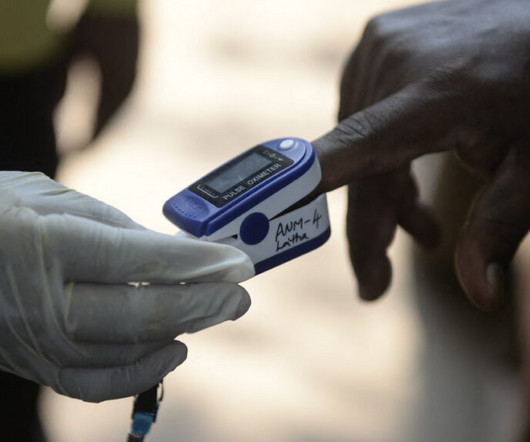
The new research analyzed paperwork submitted for FDA approval to market a device for 767 oximeters that had been approved between 1978-2024 and had accessible information about performance testing.
Schwartz — FDA recently published a Federal Register (FR) Notice [ Docket No. FDA-2024-N-3945 ] announcing the publication of a draft strategy document, for public comment, outlining specific actions FDA plans to take to facilitate the use of innovative manufacturing technologies.
FDA inspections Identification of data integrity deviations Of the 70 Warning Letters issued by the US Food and Drug Administration (FDA) so far in 2024, three have identified data integrity issues at pharmaceutical manufacturing sites outside the US. In a letter issued to China-based Sichuan Deebio Pharmaceutical Co.
Draft guidance published by the US Food and Drug Administration (FDA) in December 2023, discussed quality considerations for topical ophthalmic drug products, including key considerations for extractables and leachables (E&L) testing. This document is revised from a version published in October 2023.
Walsh — Among FDA-regulated establishments and stakeholders, there is one word that makes everyone go on edge – the dreaded FDA “inspection.” In FY2023, FDA conducted over 1000 inspections under the BIMO program. We also detail some of our recommended best practices to achieve success when FDA comes knocking.
antitrust regulators into pharmaceutical industry middlemen has been stymied because the companies have failed to provide many of the documents that, in some cases, were requested as far back as June 2022 , STAT writes. The … A much-anticipated inquiry by U.S.
The FDA may have safety concerns abut bluebird bio’s gene therapy for rare, fatal disease cerebral adrenoleukodystrophy (CALD), but its advisors believe its benefits far outweigh the risks. bluebird is also conducting the phase 3 ALD-104 trial of eli-cel in CALD, which is due to generate results in 2024.
Our 2024 nursing calculations quiz is now available. Food and Drug Administration (FDA) receives more than 100,000 reports associated with a suspected medication error. Since medications are usually dosed milligrams per kilogram, learn to consistently convert and document the patient’s weight in kilograms. Each year, the U.S.
By Sara M Keup In reviewing some new 2025 pharmacy rules/laws a couple interesting changes caught this bloggers attention: As of December 30, 2024, Missouri has rolled out new rules related to prescription drug delivery requirements. The California Board of Pharmacy updated many of the application forms and guidelines in late 2024.
Gibbs & Ana Loloei & Véronique Li, Senior Medical Device Regulation Expert — FDA has long touted the use of real-world evidence ( RWE ). FDA recognizes the potential of RWE to support regulatory submissions of medical devices and to inform benefit-risk analysis of such products, while assuring patients have timely access to devices.
Can the FDA Keep them Safe”—focuses on FDA’s role in regulating clinical applications of AI/ML technologies and the impact of FDA’s regulatory requirements on the type of products developers choose to bring to market and how quickly they can be improved once there. demographic bias of data fed to algorithms).
Lenz, Principal Medical Device Regulation Expert — FDA recently issued a draft guidance which would update the agency’s Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions guidance. FDA provides examples of changes that are likely and unlikely to impact cybersecurity of an existing device.
The primary vehicle for FDA to request cybersecurity information in premarket submissions has been guidance documents. The primary vehicle for FDA to request cybersecurity information in premarket submissions has been guidance documents.
Livornese — FDA issued a Federal Register notice on September 13, 2024, seeking feedback on the Integrated Review format that the Center for Drug Evaluation and Research (CDER) began using as part of its New Drugs Regulatory Program (NDRP) modernization effort several years ago. 13, 2024). By Deborah L.
Mullen — More than five years after FDA first announced its plan to harmonize 21 CFR Part 820 with ISO 13485, on February 2, 2024, FDA finally issued the Quality Management System Regulation (QMSR) Final Rule. FDA further retained some definitions in the QSMR. The new § 820.10 Revised § 820.3
The medication is available in both brand-name and compounded forms, although the compounded forms are not FDA approved. Some insurance plans may cover Ozempic for managing Type 2 diabetes , which is an indication that is approved by the Food and Drug Administration (FDA) to treat. Does insurance cover Ozempic?
This is the third withdrawal of an accelerated approval FDA has performed, following Avastin in 2011 and Makena in 2023. Without commenting on the merits of the decision, it is an interesting window into how FDA may use these new procedures in practice. Tobolowsky & Michelle L.
The document by the Medicines and Healthcare products Regulatory Agency (MHRA), US Food and Drug Administration (FDA) and Health Canada, contains key guidance on predetermined change control plans (PCCPs) for MLMDs. MHRA announced that it will publish its guidance in 2024.
Mullen — On June 2, 2023, FDA issued the latest version of its guidance on Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program. The document provides minor updates on procedures, incorporating recent developments and experiences in how FDA interacts with industry.
In 2012, the FDCA was modified to allow the submission of a De Novo request without the need for a prior 510(k), and set a target review time by FDA of 120 days. For example, FDA granted seven De Novos for COVID-19 related indications for use. In effect, these documents serve as road signs helping to direct new market entrants.
As a reminder, a PCCP is a document submitted in a marketing application that describes future modifications to a device that would typically require submission of a subsequent application and a description of how the sponsor will verify and validate the modified device. Draft Guidance at 4. Change Guidance at 8.
Small Dispensers and the 6/12/2024FDA Announcement Concerning DSCSA This week’s article on the recent DSCSA postponement comes from PRS Pharmacy Services. If the FDA or another regulator requests DSCSA transaction data, the DSCSA regulations give you up to 48 hours to respond. The original article can be found HERE.
Latuda (lurasidone) is a brand-name prescription drug that’s approved by the Food and Drug Administration (FDA) to treat schizophrenia as well as bipolar depression associated with bipolar I disorder. In 2019, the FDA approved a generic version of Latuda (lurasidone) , which just became available in 2023.
In the US, botanical dietary supplements can be sold without US Food and Drug Administration (FDA) approval, 1 prompting many vendors to promote the use of botanical products as dietary supplements , 2 rather than pursue a path of regulatory approval. 3 Is two too few? 3 Is two too few?
Submit proposal documents by email to ELP Proposal Submissions by September 5, 2023 at 12 PM ET. As a former participant, it was beneficial to me to engage with companies in person at their place of business and see the equipment used in manufacturing the medical devices that are reviewed by FDA. Identify the site visit duration.
4, 2024), at 1. The parties have noticed their intentions to offer into evidence documents they identified in their prehearing statements and must serve each other with a copy of the documents noticed in their prehearing statements no later than January 3, 2025. Prehearing Ruling (Dec.
156, as added by the 1984 Hatch-Waxman Amendments, for certain FDA-regulated products. The use of a surrogate endpoint can considerably shorten the time required prior to receiving FDA approval. If the confirmatory trial shows that the drug actually provides a clinical benefit, then the FDA grants traditional approval for the drug.
It can be prescribed in combination with methotrexate or as a monotherapy if the patient isn’t able to receive methotrexate, according to the final appraisal document (FAD). Gilead will receive royalties on European sales from 2024, and retains commercial rights elsewhere.
When the Food and Drug Administration (FDA) approved Mounjaro (tirzepatide) in 2022 to treat Type 2 diabetes, some people hailed the GIP and GLP-1 medication as the next great antidiabetic drug. Important note: Like Mounjaro, many of these drugs are in demand—and as of early 2024, some are in short supply, so they could be difficult to get.
While DSCSA (the Drug Supply Chain Security Act) enforcement is postponed until November 2024, it is critical to move forward in becoming compliant sooner rather than later. This record-keeping includes detailed documentation of the product’s history and movement through the supply chain. To achieve this, the U.S.
DEA recently revoked the registration of Coconut Grove Pharmacy (“Coconut Grove”), like Gulf Med Pharmacy also in Florida, for its failure to resolve prescribing red flags and document such resolution. 50,372 , 50,377 (June 13, 2024). Coconut Grove Pharmacy; Decision and Order, 89 Fed. 50,372 at 50,372-73.
Attendees were invited to submit questions to the FDA representatives. Please note that views expressed by the panelists are not necessarily representative of the position of the FDA, and that questions and responses are lightly edited for clarity. How will the FDA use Annex 1 in its final version?
Q13 Development Timeline The process of drafting the new guidance document was initiated in earnest in November 2018 when the concept paper 2 and business plan were endorsed. Since then, agencies have begun officially adopting the guidance, with the FDA doing so in March 2023.
The FDA has approved these devices for mild to moderate hearing loss. In 2024, the household income limit to qualify is under $27,180 for an individual or under $36,620 for a two-person household. In 2024, this is $30,120 for an individual and $40,880 for a couple. There are a few options to consider.
Despite the removal of some of the most controversial proposals, the final rule still contains a variety of significant changes, which become effective on November 19, 2024. Yet another rejection was a proposal requiring a diagnosis on Medicaid prescriptions as a condition for claims payment. The most noteworthy are the following: 1.
Initial appointments to both the Board and advisory council will occur by January 1, 2024. Prescription Drug Affordability Board The law also establishes a nine-member Prescription Drug Affordability Board and an 18-member stakeholder advisory council to provide advice to the Board on drug cost issues.
USA The FDA established the Emerging Technology Program (ETP) in 2014 and has actively promoted the program 8. Results from each part will be presented periodically at ISPE meetings throughout 2023 and 2024. www.fda.gov/about-fda/oncology-center-excellence/project-orbis. 22 June 2023. 22 June 2023. EMA/321483/2020. 3 July 2020.
To encourage the development of such guidance, Congress directed FDA to issue a draft guidance not later than 12 months after the date of FDORA’s enactment and to finalize such guidance not later than 9 months after closing the comment period. intended use population. population in light of the U.S. role in global health.
Questions and Answers Regarding Food Allergens, Including the Food Allergen Labeling Requirements of the Federal Food, Drug, and Cosmetic Act This final guidance replaces previous draft and final guidance documents on food allergen labeling that FDA issued in November 2022, which we discussed in a previous post. By Sophia R.
According to the legal documents , the plaintiffs accused the companies of “violating federal antitrust laws, alleging “per se” and “rule of reason” violations”. Gilenya is an oral medication for multiple sclerosis. Fighting Entresto generics.
Farquhar This is the first in a series of blog posts on tips for successfully handling an FDA inspection. Using publicly available examples, these lessons will illustrate potential pitfalls and strategies for interacting with FDA during and after an inspection. FD&C Act 501(j).
Valentine We recently blogged about a new December 2024 draft guidance about accelerated approval (the December 2024 draft guidance). FDAs withdrawal authority when a confirmatory trial is not conducted with due diligence was expanded to include that FDA could specify the conditions for a postapproval study.
The passing of the law which, among other things, amended the Federal Food, Drug, and Cosmetic Act to include requirements for facility registration and product listing, safety substantiation, adverse event reporting, and requirements for FDA to issue several new regulations, brought excitement and anxiety. Just on the cusp of 2024, on Dec.
Livornese & JP Ellison — On November 8, 2024, FDA issued a proposed order to remove the oral decongestant ingredient phenylephrine (including both phenylephrine hydrochloride and phenylephrine bitartrate) (collectively, PE) from the OTC monograph on the basis of a lack of effectiveness. By Deborah L.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.

















































Let's personalize your content