This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Karst The FDA Reduction-in-Force (Termination)or RIF(T)announced last week has resulted in countless stories in the press and on personal LinkedIn accounts from those RIFd. As folks steeped in the world of generic drugs And Hatch-Waxman know, theres a lot that happens before FDA can take action on an ANDA.
FDA inspections Identification of data integrity deviations Of the 70 Warning Letters issued by the US Food and Drug Administration (FDA) so far in 2024, three have identified data integrity issues at pharmaceutical manufacturing sites outside the US. In a letter issued to China-based Sichuan Deebio Pharmaceutical Co.
Lewis, Senior Regulatory Device & Biologics Expert — We were preparing this blogpost about FDA’s draft guidance on “Remote Interactive Evaluations” when we learned something. Compared with the COVID-centered version of the document released in April 2021, there is very little that is new or different.
Food and Drug Administration (FDA) issued two guidance documents, one draft and one final, on food allergen labeling requirements. In addition to providing additional examples of tree nuts, the draft guidance states that FDA considers the following categories of fish to be major food allergens under Section 201(qq): Jawless fish (e.g.,
At issue is a trial that Amgen is relying upon to win final approval for Lumakras, which the FDA approved on a conditional basis in 2021. But ahead of an advisory panel meeting on Thursday, the agency released documents showing its staff found “potential systemic bias” in the trial.
Pfizer has asked the FDA for an emergency authorisation of the vaccine in the US and the US government expects to the first tranche to include about 6.4 In a separate announcement, rival vaccine firm Moderna said it expected to have between 100 and 125 million doses available globally in the first quarter of 2021. million doses.
FDA , Petitioners, a liquid nicotine manufacturer, sued FDA arguing that the Agency was arbitrary and capricious in rejecting the Petitioner’s Premarket Tobacco Application (“PMTA”) in violation of the Administrative Procedure Act (“APA”). Youth behavioral data was not specifically required but FDA encouraged such information.
After a firm submits a 510(k) to FDA, FDA will request still more information after a first-pass review. According to the 2 nd Quarter FY2023 MDUFA V Performance Report , FDA issued a request for additional information (AI request) on the first FDA review cycle for 63% to 68% of 510(k)s submitted in FY2018 to FY2022.
Memorizing 45+ page document is certainly not a reasonable expectation, but one can certainly walk away with an awareness of general concepts and themes which are relevant. ” Labeling the use of an antibiotic as inappropriate or appropriate cannot simply be done based upon whether it is FDA-approved for a given indication.
The downfall of Aduhelm, the first new treatment for Alzheimer’s disease in two decades, is largely the story of a drug company choosing to maximize its potential profits at the expense of patients and taxpayers, according to a congressional investigation that cites thousands of pages of internal Biogen documents.
Prescribing Red Flags The government alleged that from at least 2017 to April 2021 Defendants knowingly filled controlled substance prescriptions “that raised obvious ’red flags’ of potential abuse or diversion.” If the pharmacist can resolve it, they must make a record of the resolution. Complaint ¶ 55.
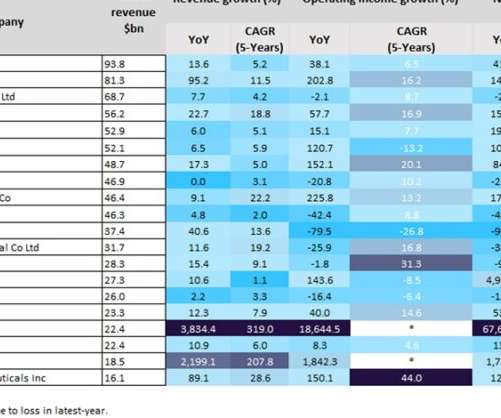
The top 13 players reported more than 10% revenue growth, with BioNTech (3,834.4%), Moderna (2,199.1%), Pfizer (95.2%) and Regeneron Pharmaceuticals (89.1%) reporting a more than 80% year-on-year (YoY) revenue growth from 2020 to 2021, according to GlobalData’s Pharma Intelligence Centre Companies Database. AbbVie reported a 22.7%
The document by the Medicines and Healthcare products Regulatory Agency (MHRA), US Food and Drug Administration (FDA) and Health Canada, contains key guidance on predetermined change control plans (PCCPs) for MLMDs.
Mullen — With comments due on the proposed LDT rule last week, FDA wasted no time updating the Unified Agenda to indicate that a final rule may be published in April ( here ). Under the Administrative Procedure Act, FDA is obligated to address major substantive issues when – and not so much if – it publishes a final rule.
This is the third withdrawal of an accelerated approval FDA has performed, following Avastin in 2011 and Makena in 2023. Without commenting on the merits of the decision, it is an interesting window into how FDA may use these new procedures in practice. Tobolowsky & Michelle L.
The announcement is no surprise as earlier this week, Pfizer’s CEO Albert Bourla said the company was preparing to file phase 3 data from the vaccine known as BNT162b2 with the FDA after gathering the required amount of safety data. billion doses by the end of 2021. Feature image courtesy of NIAID/Rocky Mountain Laboratories.
Javitt — FDA recently published a long-awaited draft guidance aimed at reducing the need for prior FDA authorization of modifications to artificial intelligence/machine learning (AI/ML)-enabled device software functions (ML-DSFs). a) , and related guidance documents (e.g., See 21 CFR 807.81(a)(3) a)(3) and 21 CFR 814.39(a)
On October 17, 2022, FDA published the list of CDRH proposed guidances for FY 2023 (see here ). These are documents on the A-list, a list of prioritized documents that FDA intends to publish during FY2023. FDA published draft guidances for these topics in December 2021 ( here and here ).
While the FDA seems to remain sceptical about the clinical benefits of Ardelyx’ chronic kidney disease (CKD) therapy Xphozah – having rejected its marketing application in 2021 – its advisors are more confident abut the drug. The post FDA panel backs Ardelyx CKD drug, despite agency concerns appeared first on.
The sampling strategy must be supported by sound and properly cited sources whose conclusions must be presented as supportive elements in the study documents. The following four elements of sampling methodology were found to be under-documented in RMM effectiveness studies: Supporting documentation for country/region selection.
The US Food and Drug Administration (FDA) has accepted Accord BioPharma’s Biologics Licence Application (BLA) for HLX02 (a proposed trastuzumab biosimilar) to treat HER2 cancer types. Accord BioPharma obtained exclusive rights from Henlius in 2021 for the development and commercialisation of HLX02 in Canada and the US.
A True Copy is an exact copy of original documentation that preserves the same content, meaning and attributes of the original. It is an electronic copy maintained in an electronic document management system. This negates the requirement for storage of paper evidence and correct GMP document management of these hardcopy evidences.
Biogen will have to make the case for its amyotrophic lateral sclerosis (ALS) therapy tofersen to an FDA advisory committee in March before it gets a decision on approval. The FDA started reviewing tofersen in July under its accelerated approval pathway, with a priority review that reflects the continued lack of effective therapies for ALS.
The US Food and Drug Administration (FDA) has released the fourth and final chapter in a series of guidance documents designed to support patient-focused drug development. The guidance also touched upon clinical trial design elements that must be explained, described, or rationalised to the FDA.
At this year’s Reuters Pharma Clinical 2021, I joined over 500 global leaders in clinical research as they discussed many of these issues under the theme, “rebuild clinical trials in the image of patient need.”. How do we increase the diversity of clinical trials?
Still, in 2021, we have seen the industry evolve and develop strategies on how to establish a viable ECA. The industry has looked to regulators for guidance, and the Food and Drug Administration (FDA) has been quick to respond. To be clear, there will always be challenges in drug development, and stakeholders are no stranger to this.
The company’s lead candidate Tenapanor is a targeted, small molecule therapy currently under FDA review. The company’s lead candidate, Tenapanor, is a targeted, small molecule therapy under FDA review for the control of serum phosphorus in adult patients with chronic kidney disease (CKD) on dialysis.
In August 2021, MSD was able to convert Keytruda’s accelerated approval for its use in in patients with locally advanced or metastatic urothelial carcinoma who are not eligible for any platinum-based chemotherapy, into a full approval. GlobalData is the parent company of Pharmaceutical Technology.
eSTAR employs targeted questions to collect specific data and information and includes applicable links to regulations, relevant guidance documents, and other resources. This has the potential to make the FDA’s review efficient as relevant information is collected upfront. printing to PDF) that states comments will not be sent to FDA.
This blog provides an update on the DHT-related PDUFA VII goals that were targeted for completion in the first two quarters of FDA’s Fiscal Year (FY) 2023, including: By the end of Q2 FY 2023, FDA will establish a DHT framework document to guide the use of DHT-derived data in regulatory decision-makings for drugs and biological products.
The diagnostic and testing companies have been serving as a pivotal foundation stone of the healthcare sector With a global market value of $165.58B in 2021, the diagnostic and testing market size is anticipated to touch the threshold of $348.75 Varex’s 2022 diagnostic segment revenue increased by 4.69% as compared to 2021.
Mullen — As of October 1, 2023, all 510(k) submissions, unless exempted, must be submitted to FDA using the electronic Submission Template And Resource ( eSTAR ). Currently, eSTAR is voluntary for medical device De Novo submissions, but FDA has initiated the process of requiring De Novos to be submitted using eSTAR.
According to the legal documents , the plaintiffs accused the companies of “violating federal antitrust laws, alleging “per se” and “rule of reason” violations”. 2021 was a year of continued innovation and change in the Biopharmaceutical industry. Gilenya is an oral medication for multiple sclerosis. 10,633,344.
156, as added by the 1984 Hatch-Waxman Amendments, for certain FDA-regulated products. The use of a surrogate endpoint can considerably shorten the time required prior to receiving FDA approval. If the confirmatory trial shows that the drug actually provides a clinical benefit, then the FDA grants traditional approval for the drug.
Lewis, Senior Regulatory Device & Biologics Expert — On October 20, 2023, FDA announced the availability of the final guidance authored by CBER titled “Voluntary Consensus Standards Recognition Program for Regenerative Medicine Therapies.” Existing published VCS may be identified internally by FDA or externally by stakeholders.
FDA has been implementing the UDI system with different compliance dates for different types of medical devices to ensure a smooth implementation. The compliance dates were first published in 2013, and subsequently updated in various guidance documents and regulations published by FDA. See 21 CFR 801.40(d).
Improvements have been observed by various regulatory agencies in Latin America for the acceptance and implementation of international standards—for instance, the ICH Common Technical Document (CTD) format. Additional required information beyond ICH guidelines, or non-value-added documents (e.g., 6, and 2.3.S.7 7 are required.
If the complaint is upheld, Enanta could be in line for a share of Pfizer’s revenues from Paxlovid, expected to make a colossal $22 billion in sales this year, after launching in December 2021.
Jörg Zimmermann, International Board Chair for 2021–2022 and Vice President, Vetter Development Service, External Affairs, Vetter Pharma-Fertigung GmbH & Co., ISPE continues to build on its mission and commitment to providing content for the industry through guidance documents and its CoPs, including expanding CoP topics. Past Chair.
The FDA had approved the drug for use as UC treatment in the US in May 2021. A lifelong condition, UC results in inflamed and ulcerated rectal and colonic lining. However, such biological treatments often come with serious side effects, including deep vein thrombosis (DVT).
The US Food and Drug Administration (FDA) requires that manufacturers establish whether nitrosamines, including NDSRIs, could be present in active pharmaceutical ingredients (APIs) and drug products, using the “three-step mitigation strategy described in the agency’s guidance”. Updated 24 February 2021. 18 November 2021.
Food and Drug Administration (FDA) certifies the Red Dye 40 as safe for human consumption. Apart from Red Dye 40, there are eight other certified color additives approved for use in food by the FDA: Blue No. The FDA has denied any link between the two for years. The controversy surrounding Red Dye 40 The U.S. 1, Blue No.
According to a 2021 systematic review and meta-analysis , the combination of L-theanine and caffeine has positive effects on short-term sustained attention and overall cognition. That said, the effects of caffeine are well-documented. According to the U.S.
Biogen has invested heavily in its Alzheimer’s candidate aducanumab, but an FDA advisory committee was unimpressed with the company’s rehashed data for the anti-amyloid drug and voted that it didn’t support efficacy. The post Biogen bulks up pipeline with Sage drugs in $3bn deal appeared first on.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.


















































Let's personalize your content