This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
A study of the causes of warning letters issued by the US Food and Drug Administration (FDA)’s Center for Drug Evaluation and Research (CDER) and Center for Devices and Radiological Health (CDRH) between 2010 and 2020 revealed that poor current good manufacturing practice (cGMP) compliance and misbranding were the most common citations.
The US Food and Drug Administration (FDA) has released a new draft guidance to further support the use of decentralised clinical trials (DCTs) for drugs, biologics and devices. This draft guidance builds on recommendations published by the agency in 2020.
On an average, around 50 drugs are approved by the US Food and Drug Administration (US FDA) annually. This continuously growing pipeline of pharmaceutical drug products has inadvertently led to an increase in the demand for their associated primary packaging and Secondary Packaging solutions. . trillion in 2023.
The NDA for Abicipar pegol, which utilizes DARPin technology was rejected by the FDA secondary to concerns of inflammation with the 2 mg dose”. What current topic will you be addressing in your presentation, and what would you say makes it relevant to 2020? “We 23 rd -24 th November 2020. Ophthalmic Drugs Conference.
After a firm submits a 510(k) to FDA, FDA will request still more information after a first-pass review. According to the 2 nd Quarter FY2023 MDUFA V Performance Report , FDA issued a request for additional information (AI request) on the first FDA review cycle for 63% to 68% of 510(k)s submitted in FY2018 to FY2022.
Valentine — On November 22, 2022, FDA approved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
billion since 2020. It will boost the site’s its capacity for parenteral filling, device assembly and packaging even further. The plant in Research Triangle Park has already received hefty investment from Lilly, and the new tranche will take the tally to $1.7
Already scrambling to refile its obeticholic acid (OCA) drug for non-alcoholic steatohepatitis (NASH) after the FDA rejected it last year, Intercept Pharma was hoping for more luck in Europe, but has now withdrawn its EU marketing application. Shares in Intercept slipped around 15% after it announced the news.
2022, FDA published a draft guidance on FDA’s implementation of the Over-the-Counter Monograph Drug User Fee Program (OMUFA). FDA is authorized to charge an annual facility fee for OTC monograph drug facilities. FDA will not refund any fees that have been incurred already). By Riëtte van Laack & Deborah L.
By Riëtte van Laack — On September 28, 2022, FDA announced the availability of the proposed rule for the implied nutrient content claim “healthy.” The term healthy, as an implied nutrient claim, was first defined by FDA in 1994. FDA also announced it would be re-evaluating the regulatory criteria for use of the “healthy” claim.
It can also treat parasitic infections, like giardia, and is frequently used off-label (for a non-FDA-approved use) to help treat Crohn’s disease, bite wounds, and oral infections. Additionally, i n the case of bacterial vaginosis, this may improve your outcome, per a 2020 study.
Check the drug’s packaging and label carefully before giving it to your pet. The FDA says Zyrtec’s effects typically last around 24 hours for humans, and studies show that its elimination half-life is around 8.3 Important Note: While standard Zyrtec is safe for dogs , never give a dog Zyrtec-D. How quickly does Zyrtec work in dogs?
Always follow the directions on the packaging or consult a healthcare professional if you have concerns about the appropriate dosage. In addition, the FDA label for the nighttime forms of Mucinex containing acetaminophen recommends avoiding three or more drinks per day.
As does the US Food and Drug Administration (FDA): REMS Assessment: Planning and Reporting Guidance for Industry Draft Guidance. Patient-Focused Drug Development: Collecting Comprehensive and Representative Input Guidance for Industry, Food and Drug Administration Staff, and Other Stakeholders; June 2020. Available from: [link].
There’s currently no generic alternative for Stelara, but in 2023, the Food and Drug Administration (FDA) approved a biosimilar substitute called Wezlana. Getting a drug approved by the FDA is a very expensive process,” says rheumatologist Stella Bard, MD. In October 2023, the FDA approved Wezlana, a biosimilar for Stelara.
For people with Type 2 diabetes, Ozempic is a game-changer: This FDA-approved drug not only lowers blood sugar but can also help people lose weight. American Diabetes Association (undated) Achieving Type 2 diabetes remission through weight loss , National Institute of Diabetes and Digestive and Kidney Diseases (2020) Carbohydrates.
If a patient wants to report an adverse reaction to the FDA, they need to fill out Form 3500B. On the other hand, if medication is dispensed in the drug company’s original packaging, the law requires an expiration date along with lot number and NDC number ( StatPearls , May 1, 2023 ). The FDA does virtually no testing.
Zyrtec was approved by the United States Food and Drug Administration (FDA) in 1995. The first generic of prescription Zyrtec, cetirizine, was approved by the FDA in 2008, around the time the medication became available over the counter. For example, one 2020 study from Taiwan compared brand-name and generic antidepressants.
new combined oral contraceptive was approved by the FDA (Nextstellis®) in April 2021. In conclusion, Nextstellis is a recent FDA-approved oral contraceptive that offers patients another alternative for birth control. Nextstellis (Drospirenone and Estetrol) [package insert]. Published April 7, 2020. 2020, [link].
Of the 175 new drugs approved by the US Food and Drug Administration (FDA) between 2016 and 2019, most were biologics. The payload is packaged inside a dissolvable needle, loaded within a microsyringe that is attached to a folded, self-inflating balloon. Biologics represent an increasingly large part of the pharmaceutical industry.
Four factor prothrombin complex concentrate (4F-PCC) is FDA approved to reverse vitamin K antagonists (VKAs) such as warfarin in adult patients with acute major bleeding or a need for an urgent surgery/invasive procedure. Dosing According to Package Insert. Kcentra (prothrombin complex concentrate [Human]) [package insert].
The FDA, PIC/S, and WHO have all emphasized the importance and benefits of data flows in their guidance on data integrity. The goal is to break down complex processes into easy-to-grasp packages, including data privacy and information technology (IT) security. In the fourth and last step, the defined work packages are executed.
It’s now globally accessible due to Gilead’s work with a network of generic manufacturers and their ability to sign licensing agreements just days after the US FDA registered its authorisation. If you get past the regulatory hurdles, then you have to look at labelling requirements, packaging requirements, and quality assurance requirements.”.
Follow these steps to try nasal irrigation for congestion: Prepare the nasal rinse solution by mixing saline with distilled water per the package instructions. The FDA recently announced that the oral version of phenylephrine, a popular ingredient in some decongestants, is ineffective. John Hopkins Medicine Turmeric.
Many countries have introduced serialisation legislation which requires product identifiers to be affixed to each package to provide traceability throughout the distribution supply chain. However, traceability and security measures focused on the packaging level may not be enough to protect patients.
You’ve probably eaten seaweed as part of your favorite sushi dish before or even snacked on a package of dried seaweed from the shelf of your local health food store. A 2020 study suggests that a daily intake of three to four grams of dried sea moss is likely safe for healthy adults.
Safety measures while using CoQ10 What are the FDA warnings about CoQ10? The Food and Drug Administration (FDA) has not issued any warnings about CoQ10. It is available over the counter, so the FDA has determined that it’s safe to use without medical supervision. Who should never take CoQ10? Who should use caution with CoQ10?
The US FDA’s review is expected to be completed in H2’23 Under the Aug 2020 agreement, Alvotech and Teva collaborated for the exclusive commercialization in the US of five biosimilar product candidates incl. chronic plaque psoriasis in the US & 9 other countries incl.
In addition to the COR, some Latin American markets require approval in the COO, which is defined as the country where the drug is manufactured, packaged, or exported from. Sometimes, COAs for the drug substance, the excipients used in the formulation, and the primary and nonfunctional secondary packaging material are requested.
As part of the US Food and Drug Administration (FDA) QbD pilot 4 in 2005–2006, the need to convey how the control strategy is linked to the target product profile (TPP) and quality target product profile (QTPP) was discussed. Direct feedback from the US FDA on this pilot is shared in the discussion.
As part of the US Food and Drug Administration (FDA) QbD pilot 4 in 2005–2006, the need to convey how the control strategy is linked to the target product profile (TPP) and quality target product profile (QTPP) was discussed. Direct feedback from the US FDA on this pilot is shared in the discussion.
m/s recommendation made its way into the US Food and Drug Administration (FDA)’s “Sterile Drug Products Produced by Aseptic Processing—Current Good Manufacturing Practice” 7 and into the EC GMP Annex 1 “Manufacture of Sterile Medicinal Products” in 2003. x, a free and open-source computational fluid dynamics (CFD) software package.
Why, then, are FDA inspections abroad so infrequent and disappointing? FDA Inspections Suspended: During the COVID pandemic, the Food and Drug Administration suspended its inspections at most foreign pharmaceutical facilities. Even under the best of conditions, the FDA never inspects all foreign pharmaceutical plants annually.
Amidst numerous warning letters distributed by the US Food and Drug Administration (FDA) to pharmaceutical manufacturers in recent months, in July this year, the US regulatory body published its analysis on drug product quality in 2022. Eight FDA warning letters were handed to European companies in 2018. Edge Biologicals Inc.,
Mallinckrodt has finally claimed FDA approval for terlipressin as a treatment for hepatorenal syndrome (HRS), after manufacturing problems scuppered an earlier attempt. The drug is already approved for this indication in dozens of other countries around the world, including much of Europe, Australia and New Zealand.
The FDA has issued a complete response letter to Mallinckrodt for terlipressin, on the grounds that the company had been forced to change its packaging and labelling manufacturing facility for the drug. The post FDA knocks back Mallinckrodt’s kidney drug terlipressin appeared first on.
For changes requiring approval before implementation, once the necessary information has been submitted across regions, each health authority must review the data package and documentation based on local requirements. The European Medicines Agency (EMA) issued its implementation guidance in March 2020.
For example, a recent 2020 study found that less than 25% of the 50 testosterone supplements evaluated had scientific research backing their positive effects on testosterone (even though nearly all of them—90%—purported to boost T). Food and Drug Administration ( FDA ), so you may be taking a risk when you choose to add one to your diet.
Manufacturing problems wreaked havoc with Mallinckrodt’s first attempt to secure FDA approval of its treatment candidate terlipressin for hepatorenal syndrome (HRS), but the drugmaker is having another attempt. There is no FDA-approved therapy available. billion settlement in 2020 which was finally accepted earlier this year.
Only 25% of API production for generic drugs took place in Europe in 2020, according to a November 2022 Medicines for Europe report. The European Commission plans to launch its revised EU pharmaceutical package in Q1 2023 to ease drug shortages,” she says. For innovative medicines, this share was 77%.
Large (market cap $10–100bn) and mega-cap (market cap >$100bn) sponsors also require contract manufacturers and packagers with specialist injectable capabilities in the case of cell and gene therapies, as shown in the report titled Contract Injectable Packaging Trends in the Bio/Pharma Industry (August 2022).
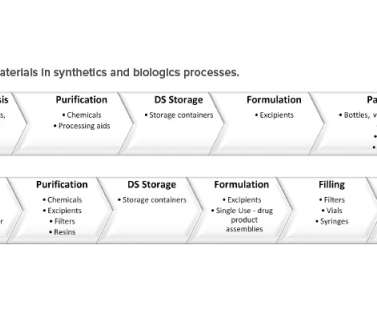
In this article, the term “raw material” refers to a material used in the manufacturing and packaging of a drug substance (DS) or a drug product (DP). Finally, the DP is packaged in a suitable container to ensure continued quality. It is considered a moderate change by the FDA (CBE30) and NMPA.
“However, since children’s dosing recommendations may vary between Benadryl products, it’s best to check the package label for dosing instructions to ensure appropriate and safe dosing,” says Dr. Johnson-Arbor. Dosing for the topical versions of Benadryl will differ, so always follow package instructions or consult your healthcare provider.
million in 2020 and with the rapid growth of the industry, is expected to grow at a CAGR of 8.95% up to 2027. Guest FDA Speaker: John Barr Weiner, Associate Director for Policy and Product Classification Office, Office of Combination Products FDA. . Primary Packaging material designers. Secondary packagers.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.














































Let's personalize your content