This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The US Food and Drug Administration (FDA) has sent a warning letter to KVK-Techs drug manufacturing facility following an inspection in April 2019. The document identified significant violations of current good manufacturing practice (cGMP) regulations for finished pharmaceuticals. Whats next for KVK-Tech?
In March 2020, as Bauman was preparing to undergo the surgery at a Mount Sinai medical facility in midtown Manhattan, he said he was invited to participate in a research study — one only open to patients already committed to undergo DBS at Mount Sinai. The review, which lists consultations with 16 different FDA officials, was scathing.
A study of the causes of warning letters issued by the US Food and Drug Administration (FDA)’s Center for Drug Evaluation and Research (CDER) and Center for Devices and Radiological Health (CDRH) between 2010 and 2020 revealed that poor current good manufacturing practice (cGMP) compliance and misbranding were the most common citations.
FDA advisors will scrutinise three cancer immunotherapies granted conditional approvals at a three-day meeting this week, to see if they should stay on the market. . Briefing documents published by the FDA ahead of the meeting suggest that discussion will focus on ongoing trials that may serve as alternative confirmatory studies.
On February 7, at a town hall organised to discuss clinical trial designs for gene therapies, FDA experts pushed pharma players to look for ways to establish clinical effectiveness despite the challenges in recruiting patients with rare diseases.
FDA , Petitioners, a liquid nicotine manufacturer, sued FDA arguing that the Agency was arbitrary and capricious in rejecting the Petitioner’s Premarket Tobacco Application (“PMTA”) in violation of the Administrative Procedure Act (“APA”). Youth behavioral data was not specifically required but FDA encouraged such information.
According to press reports, Slaoui expects an influential FDA advisory committee to say today that people with severe allergies “should not take the vaccine until we know exactly what happened.”. Feature image copyright BioNTech SE 2020, all rights reserved.
After a firm submits a 510(k) to FDA, FDA will request still more information after a first-pass review. According to the 2 nd Quarter FY2023 MDUFA V Performance Report , FDA issued a request for additional information (AI request) on the first FDA review cycle for 63% to 68% of 510(k)s submitted in FY2018 to FY2022.
The announcement is no surprise as earlier this week, Pfizer’s CEO Albert Bourla said the company was preparing to file phase 3 data from the vaccine known as BNT162b2 with the FDA after gathering the required amount of safety data. billion doses by the end of 2021. Feature image courtesy of NIAID/Rocky Mountain Laboratories.
The FDA has dropped a bomb on Eli Lilly’s marketing application for cancer immunotherapy sintilimab ahead of an advisory committee meeting due to take place on Thursday. ” While ODAC could still decide to back the drug at the meeting, the FDA isn’t beholden to follow its experts’ advice. .”
The US Food and Drug Administration (FDA) has accepted Accord BioPharma’s Biologics Licence Application (BLA) for HLX02 (a proposed trastuzumab biosimilar) to treat HER2 cancer types. Accord BioPharma is the US specialty division of Intas Pharmaceuticals. HLX02 is intended for adjuvant treatment of HER2-overexpressing breast cancer.
An FDA advisory committee has recommended approval of Cidara Therapeutics’ once-weekly antifungal therapy rezafungin, setting it up to become the first new option for severe infections caused by Candida species in a decade. Melinta is privately-owned, after emerging from bankruptcy protection in 2020.
Shares in US biotech Y-mAbs Therapeutics have lost almost a third of their value after FDA advisors unanimously rejected its brain cancer therapy 131I-omburtamab in 16 to 0 vote. Changes in standard and supportive care over the time period could also have skewed the results, said the FDA reviewer.
Javitt — FDA recently published a long-awaited draft guidance aimed at reducing the need for prior FDA authorization of modifications to artificial intelligence/machine learning (AI/ML)-enabled device software functions (ML-DSFs). a) , and related guidance documents (e.g., See 21 CFR 807.81(a)(3) a)(3) and 21 CFR 814.39(a)
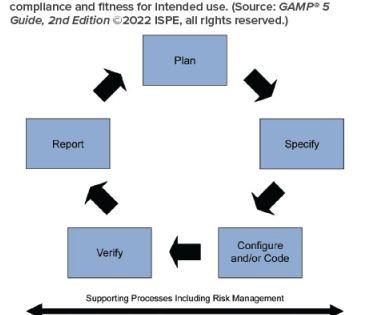
Validation projects have often been documentation-focused exercises, more befitting a grammar exercise than artifacts supportive of regulated activity. The GAMP 5 Second Edition guide emphasises that focus should be on value-adding activities rather than documentation for documentation’s sake.”
The sampling strategy must be supported by sound and properly cited sources whose conclusions must be presented as supportive elements in the study documents. The following four elements of sampling methodology were found to be under-documented in RMM effectiveness studies: Supporting documentation for country/region selection.
FDA advisors have voted against approval of Eli Lilly and Innovent Biologics’ cancer immunotherapy sintilimab, undermining hopes of a new, lower-priced option in the PD-1/PD-L1 inhibitor class. Lilly said that along with Innovent it would work with the FDA as it completes its review of the sintilimab application.
Of all products approved by the European Medicines Association (EMA) between 2017 and 2020, 68% are currently available in England, a figure that stands at 54% in Scotland. In addition, the median time between regulatory approval and the first patient receiving a first dose was 247 days in 2020 – an increase of 25 days since 2018.
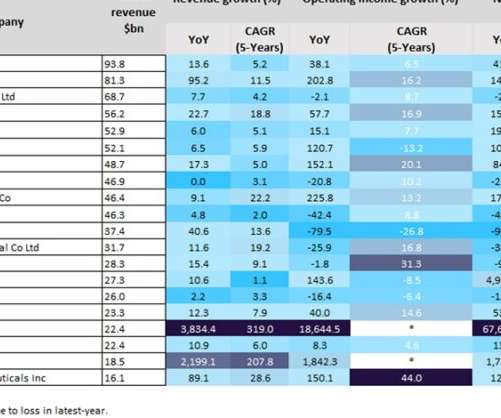
The top 13 players reported more than 10% revenue growth, with BioNTech (3,834.4%), Moderna (2,199.1%), Pfizer (95.2%) and Regeneron Pharmaceuticals (89.1%) reporting a more than 80% year-on-year (YoY) revenue growth from 2020 to 2021, according to GlobalData’s Pharma Intelligence Centre Companies Database.
In the US, botanical dietary supplements can be sold without US Food and Drug Administration (FDA) approval, 1 prompting many vendors to promote the use of botanical products as dietary supplements , 2 rather than pursue a path of regulatory approval. 3 Is two too few? 3 Is two too few?
By Riëtte van Laack — On September 28, 2022, FDA announced the availability of the proposed rule for the implied nutrient content claim “healthy.” The term healthy, as an implied nutrient claim, was first defined by FDA in 1994. FDA also announced it would be re-evaluating the regulatory criteria for use of the “healthy” claim.
12 of February 2020 again received 2,000 comments from public consultation. The first draft for comments from industry stakeholders was published in 2017 and generated 6000 comments.
156, as added by the 1984 Hatch-Waxman Amendments, for certain FDA-regulated products. The use of a surrogate endpoint can considerably shorten the time required prior to receiving FDA approval. If the confirmatory trial shows that the drug actually provides a clinical benefit, then the FDA grants traditional approval for the drug.
HHS noticed an increasing trend of illegal kickbacks in connection with speaker programs and in November 2020, the Office of Inspector General issued a Special Fraud Alert on the topic. Today, these two documents are key in navigating speaker programs in the industry. See 42 U.S.C. 1320a-7b(b).
“For example, in early 2020, we enrolled somewhere between 3,000 to 3,500 participants each week. In March 2020, when the pandemic hit, we had to pause in-person recruitment. Food and Drug Administration (FDA). But that also provided us with an unexpected finding.” About the author.
Waylivra – its second drug – was rejected by the FDA in 2018 on concerns about its safety, including the risk of serious bleeding and low blood platelet count. Fantatic news for the FCS Community – NICE has issued a positive Final Evaluation Document (FED) for Volanesorsen. — Action FCS (@ActionFCS) September 18, 2020.
Koblitz — For years , FDA has been wrestling with questions about what patents should be listed in the Orange Book, but, as we have reported, FDA has made little to no progress on addressing those questions. One of those pressing questions that remains unanswered involves the listing of REMS patents in the Orange Book.
In 2020, the Federal Trade Commission issued a report regarding settlements reached between brand and generic manufacturers in FY 2017 and noted that “for the first time since [FY] 2004, no settlement agreement in [FY] 2017 contains a no-[authorized generic] AG commitment.”. Gilenya is an oral medication for multiple sclerosis.
The FDA granted full approval to Par’s generic on 28 March, 2013. According to court documents cited by Reuters, annual sales of Exforge in the US were over $400 million before generic versions reached the market. The post Novartis agrees $245m settlement over Exforge generics delay appeared first on.
In the United States, the Food and Drug Administrator (FDA) has so far published three draft guidance documents on patient-focused drug development, with a fourth expected in the coming months. EMA and FDA backing represents a huge step forward for patient centricity as a force for good in pharmaceutical development.
The US Food and Drug Administration (FDA) requires that manufacturers establish whether nitrosamines, including NDSRIs, could be present in active pharmaceutical ingredients (APIs) and drug products, using the “three-step mitigation strategy described in the agency’s guidance”. 2020, 24 (12), 2915–292. 2020; 24(9): 1558–1585.
Food and Drug Administration (FDA) certifies the Red Dye 40 as safe for human consumption. Apart from Red Dye 40, there are eight other certified color additives approved for use in food by the FDA: Blue No. The FDA has denied any link between the two for years. The controversy surrounding Red Dye 40 The U.S. 1, Blue No.
Improvements have been observed by various regulatory agencies in Latin America for the acceptance and implementation of international standards—for instance, the ICH Common Technical Document (CTD) format. Additional required information beyond ICH guidelines, or non-value-added documents (e.g., 6, and 2.3.S.7 7 are required.
Yes, as of March 2020 , insulin is considered a biologic product by the FDA. It was first approved in June 2020. Regulatory bodies such as the FDA conduct thorough tests to confirm that they meet safety, purity, and potency standards. Is insulin a biologic medication? It was first approved in December 2021.
A lack of transparency The importance of setting the primary outcome prior to commencing a study and not deviating from the original protocol, was first highlighted in 1990 by Jay Siegel , a physician and research scientist working for the FDA in the US.
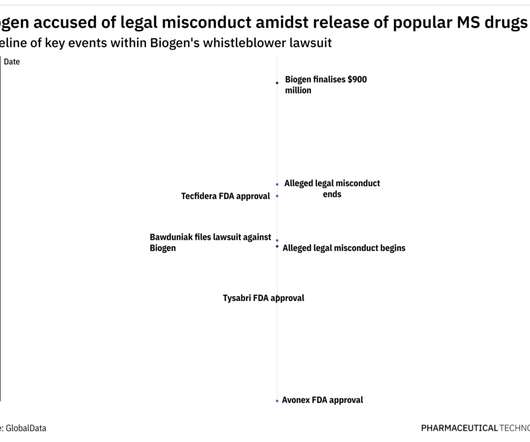
These, along with other similar claims, meant Biogen owed potential damages of $1,036,900,151 to the US and the various States, as per court documents. Tecfidera was approved in 2013 to treat MS, and in August 2020, Mylan launched its first FDA-approved dimethyl fumarate generic to the market.
Concept Paper: M4Q(R2) Common Technical Document on Quality Guideline.” As part of the US Food and Drug Administration (FDA) QbD pilot 4 in 2005–2006, the need to convey how the control strategy is linked to the target product profile (TPP) and quality target product profile (QTPP) was discussed. The authors propose using Module 2.3
Concept Paper: M4Q(R2) Common Technical Document on Quality Guideline.” As part of the US Food and Drug Administration (FDA) QbD pilot 4 in 2005–2006, the need to convey how the control strategy is linked to the target product profile (TPP) and quality target product profile (QTPP) was discussed. The authors propose using Module 2.3
Because this product was approved fairly recently in 2020 , it will be quite some time before a generic version is available. The device is being prescribed as indicated by the Food and Drug Administration (FDA). You have a history of problematic hypoglycemia with documentation of specific hypoglycemic events.
More than 100 clinical trials were initiated in Georgia from 2020 to 2021, with the majority from international sponsors and a focus on managing Phase II-III multinational trials.
The alleged counterfeit drug sales were traced back to August of 2020 and continued well into 2021. To the extent that SafeChain sold any counterfeit medication, SafeChain was an unwitting participant and notified the FDA of customer complaints as required by law. Where’s the FDA? Time will tell. . Emphasis added). .
Food and Drug Administration (FDA) approves it for treating rheumatoid arthritis , systemic lupus erythematosus, and malaria. Hydroxychloroquine also received a surge in media presence as one of the many drugs used off-label in treating coronavirus after the pandemic that started in 2020.
It was time for ISPE to update this key guidance document to reflect technological progress. Concepts of computerized software assurance (CSA) as discussed in the US FDA Center for Devices and Radiological Health (CDRH) Case for Quality program 1 are also explored and applied. 15 September 2020. 1 June 2021.
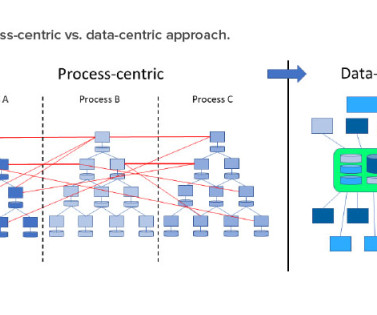
The FDA, PIC/S, and WHO have all emphasized the importance and benefits of data flows in their guidance on data integrity. 3 , 4 RAMI integrates all assets, including physical items, software, administrative shell, documents, and personnel. Many organizations already have documented process maps. Figure 3 shows all four steps.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.















































Let's personalize your content