This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
FDA advisors will scrutinise three cancer immunotherapies granted conditional approvals at a three-day meeting this week, to see if they should stay on the market. . Briefing documents published by the FDA ahead of the meeting suggest that discussion will focus on ongoing trials that may serve as alternative confirmatory studies.
Cato — On October 21, 2022, FDA published a draft guidance document titled Select Updates for the Breakthrough Devices Program Guidance: Reducing Disparities in Health and Health Care. This draft guidance proposes updates to FDA’s Breakthrough Devices Program, which is outlined in a separate December 2018 guidance document.
Gibbs & Ana Loloei & Véronique Li, Senior Medical Device Regulation Expert — FDA has long touted the use of real-world evidence ( RWE ). FDA recognizes the potential of RWE to support regulatory submissions of medical devices and to inform benefit-risk analysis of such products, while assuring patients have timely access to devices.
After a firm submits a 510(k) to FDA, FDA will request still more information after a first-pass review. According to the 2 nd Quarter FY2023 MDUFA V Performance Report , FDA issued a request for additional information (AI request) on the first FDA review cycle for 63% to 68% of 510(k)s submitted in FY2018 to FY2022.
FDA , Petitioners, a liquid nicotine manufacturer, sued FDA arguing that the Agency was arbitrary and capricious in rejecting the Petitioner’s Premarket Tobacco Application (“PMTA”) in violation of the Administrative Procedure Act (“APA”). Youth behavioral data was not specifically required but FDA encouraged such information.
Dr Stanley Cohen was sued by Alafi Capital and the Christopher Alafi Family Trust – the only investors in Nuredis – in 2018. The lawsuit claimed he misled them whist persuading them to invest $20 million in the biotech, set up to develop a candidate therapy for neurodegenerative disease Huntington’s disease , in return for a 20% stake.
Mullen — On December 9, 2022, FDA issued a draft guidance document on the Voluntary Malfunction Summary Reporting (VMSR) Program for medical devices. The purpose of this program is to reduce the volume of reports that a manufacturer needs to submit to FDA and to “make malfunction event trends more readily apparent.”.
Mullen — With comments due on the proposed LDT rule last week, FDA wasted no time updating the Unified Agenda to indicate that a final rule may be published in April ( here ). Under the Administrative Procedure Act, FDA is obligated to address major substantive issues when – and not so much if – it publishes a final rule.
Why, then, are FDA inspections abroad so infrequent and disappointing? FDA Inspections Suspended: During the COVID pandemic, the Food and Drug Administration suspended its inspections at most foreign pharmaceutical facilities. Even under the best of conditions, the FDA never inspects all foreign pharmaceutical plants annually.
Lenz, Principal Medical Device Regulation Expert — On September 6, 2023, FDA announced its latest efforts to modernize the 510(k) process, outlining FDA’s latest improvements to strengthen the 510(k) Program and announcing release of three draft guidance documents. stated, “ We appreciate the IOM’s report on the 510(k) program.
A True Copy is an exact copy of original documentation that preserves the same content, meaning and attributes of the original. It is an electronic copy maintained in an electronic document management system. This negates the requirement for storage of paper evidence and correct GMP document management of these hardcopy evidences.
Brevig, Senior Regulatory Device and Biologics Exper — FDA recently published Alternative or Streamlined Mechanisms for Complying with the Current Good Manufacturing Practice Requirements for Combination Products; List under the 21st Century Cures Act in the Federal Register (FR Notice). FDA must also review the list periodically.
The industry has looked to regulators for guidance, and the Food and Drug Administration (FDA) has been quick to respond. Within this framework, the FDA seeks to answer outstanding questions on how it will ultimately assess RWD, as well as address any prospective issues, such as cohort bias and data quality.
Waylivra – its second drug – was rejected by the FDA in 2018 on concerns about its safety, including the risk of serious bleeding and low blood platelet count. Fantatic news for the FCS Community – NICE has issued a positive Final Evaluation Document (FED) for Volanesorsen.
In 2018, over 60% of all new molecular entities came from smaller biopharma firms, compared with just over 30% in 2009. These teams will include experts in regulatory, GMP compliance and quality systems, often including ex-FDA and MHRA inspectors and industry experts.
eSTAR employs targeted questions to collect specific data and information and includes applicable links to regulations, relevant guidance documents, and other resources. This has the potential to make the FDA’s review efficient as relevant information is collected upfront. printing to PDF) that states comments will not be sent to FDA.
In addition, the median time between regulatory approval and the first patient receiving a first dose was 247 days in 2020 – an increase of 25 days since 2018. The news comes weeks after researchers found patients across Europe were already waiting longer than their American counterparts for access to new cancer drugs.
The FDA granted full approval to Par’s generic on 28 March, 2013. Plaintiffs – in this case direct purchasers, indirect purchasers and retailers including CVS Health, Kroger, Rite Aid and Walgreens Boots Alliance according to Reuters – filed suit against Novartis and Par in 2018 alleging violation of federal antitrust laws.
In the United States, the Food and Drug Administrator (FDA) has so far published three draft guidance documents on patient-focused drug development, with a fourth expected in the coming months. EMA and FDA backing represents a huge step forward for patient centricity as a force for good in pharmaceutical development.
The FDA subsequently raised serious concerns about the accuracy of testing that Theranos did conduct with its machines, and the company eventually retracted two years of blood tests. It eventually folded in 2018.
Lilly’s drug becomes an oral alternative to Sanofi and Regeneron’s biologic therapy Dupixent (dupilumab), which was recommended as a second-line option for moderate to severe atopic dermatitis in 2018. Dupilumab does not always work, and some people stop taking it because of side effects,” according to NICE’s technical appraisal document.
Food and Drug Administration (FDA) certifies the Red Dye 40 as safe for human consumption. Apart from Red Dye 40, there are eight other certified color additives approved for use in food by the FDA: Blue No. The FDA has denied any link between the two for years. The controversy surrounding Red Dye 40 The U.S. 1, Blue No.
Q13 Development Timeline The process of drafting the new guidance document was initiated in earnest in November 2018 when the concept paper 2 and business plan were endorsed. Since then, agencies have begun officially adopting the guidance, with the FDA doing so in March 2023.
Comment topics suggested by DEA include the following: Whether the rule should limit the issuance of prescriptions for controlled medications to FDA-approved indications contained in the labeling for those medications. The only schedule III-V narcotic drug that is currently approved by the FDA for OUD treatment is buprenorphine.
However, the Food and Drug Administration (FDA) didnt officially approve Basaglar as a biosimilar, but rather a follow-on insulin essentially one companys copy of another companys product. Lantus has two FDA-approved biosimilars : Semglee and Rezvoglar.
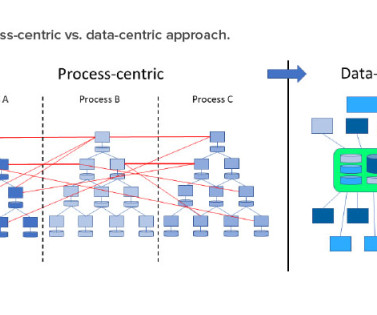
The FDA, PIC/S, and WHO have all emphasized the importance and benefits of data flows in their guidance on data integrity. 3 , 4 RAMI integrates all assets, including physical items, software, administrative shell, documents, and personnel. Many organizations already have documented process maps. Figure 3 shows all four steps.
9] A follow-up study done in 2018 (also led by Hofling) assessed the same group of 43 people six years after the clinical trial. All Lumebox products are FDA-registered, follow the Good Manufacturing Practice (GMP) system, and every batch is third-party tested. Published 2018 Nov 4. References [1] Nanan R, Wall JR.
We expect the Minnesota law to face Commerce Clause, vagueness, and possibly other constitutional challenges, similar to those brought against a generic price gouging prohibition in Maryland that was struck down by the Fourth Circuit in 2018 (see our post here ).
However, when the US Food and Drug Administration (FDA) introduced “Pharmaceutical cGMPs for the 21st Century — A Risk-Based Approach” 5 in September 2004, the paradigm shifted and these questions were asked: What is the probability that changing the airflow in a given clean area will affect the process or product in that area? 241,110 16.6
Concept Paper: M4Q(R2) Common Technical Document on Quality Guideline.” As part of the US Food and Drug Administration (FDA) QbD pilot 4 in 2005–2006, the need to convey how the control strategy is linked to the target product profile (TPP) and quality target product profile (QTPP) was discussed. The authors propose using Module 2.3
Concept Paper: M4Q(R2) Common Technical Document on Quality Guideline.” As part of the US Food and Drug Administration (FDA) QbD pilot 4 in 2005–2006, the need to convey how the control strategy is linked to the target product profile (TPP) and quality target product profile (QTPP) was discussed. The authors propose using Module 2.3
When FDA published its draft guidance on payor communications in 2017, it seemed revolutionary – introducing the concept of pre-approval information exchange for wholly unapproved products. FDA had put industry in a quandary.
FDA previously released the final guidance document in December 2018 outlining the program’s principles, features, designation criteria, and other considerations. Additionally, the 2022 draft guidance suggested the addition of a new section to the 2018 guidance on reducing disparities in health and healthcare.
The regulatory keynote was delivered by Peter Marks, MD, PhD, Director, Center for Biologics Evaluation and Research (CBER), US FDA. Marks outlined some concepts that the FDA is thinking about that could help move in this direction, such as developing a “cookbook” to standardize bespoke product development and manufacturing.
Building on these concepts, we provide insights and guidance on how to effectively support the “Human-AI-Team,” a term of the FDA 5 we believe describes very well the target operating model when interacting with AI: in a collaborative manner, complementing the strengths of cognitive as well as AI in GxP-related areas. 12 November 2018.
Cato — The long-running saga of FDA regulation of laboratory-developed tests (LDTs) has taken yet another new turn. Take for example FDA’s Unified Agenda announcement for ISO 13485 harmonization, which first occurred in the Spring of 2018 and was re-issued until a proposed rule was published in early 2022 (see our post here ).
It was usual practice for TGA to prepare additional guidance/explanatory documents for industry and for TGA inspection staff to participate in training seminars to assist industry. Paul indicated that in 2018, ICMRA introduced an Inspection Reliance Guideline which was adopted by PIC/S ( PICS Inspection Reliance Guidance ).
In the US, botanical dietary supplements can be sold without US Food and Drug Administration (FDA) approval, 1 prompting many vendors to promote the use of botanical products as dietary supplements , 2 rather than pursue a path of regulatory approval. 3 Is two too few? 3 Is two too few?
This article of mine discusses 19 thyroid-toxic products that have been banned by the FDA. Feel free to use my own health timeline as a guideline to document your health journey. 2018(1):1-10. doi: 10.1155/2018/4391579. Published 2018 Jul 2. Updated September 7, 2021. Accessed April 2022. Argunhan Z, Avci, AS.
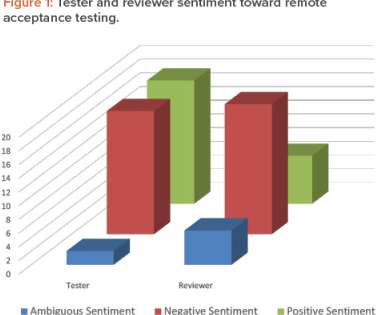
The writing of requirements, design documentation, and test scripts, and the configuration of hardware and software, can be conducted from home. During the COVID-19 pandemic, the US FDA issued guidelines for remote evaluations of drug manufacturing and bioresearch facilities. Los Angeles, CA, US: Sage Publications, 2018.
24 August 2018. Published 2018. FDA Official Warns Manufacturers of Common Problems Found in Aseptic Operations.” www.raps.org/news-and-articles/news-articles/2023/3/fda-official-warns-manufacturers-of-common-problem 25 US Food and Drug Administration Center for Drug Evaluation and Research. 6 November 2019. 7 March 2023.
It was usual practice for TGA to prepare additional guidance/explanatory documents for industry and for TGA inspection staff to participate in training seminars to assist industry. Paul indicated that in 2018, ICMRA introduced an Inspection Reliance Guideline which was adopted by PIC/S. The approach by TGA and HSA were very similar.
A 2015 case report documented the thyroid labs of a man who had recurrent hyperthyroid episodes after three of his wife’s pregnancies! September 27, 2018. Published 2018 Sep 3. Reviewed March, 2018. Like women, men can develop hormonal changes after the birth of a child, which can be linked to postpartum depression. [14]
7 A critical feature of the ETP is representation from all relevant FDA quality assessment and inspection programs, from early engagement with stakeholders through application submission and assessment. 13 An FDA-authored paper indicating support for the implementation of CM using science- and risk-based approaches followed soon thereafter.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.
















































Let's personalize your content