This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Karst The FDA Reduction-in-Force (Termination)or RIF(T)announced last week has resulted in countless stories in the press and on personal LinkedIn accounts from those RIFd. As folks steeped in the world of generic drugs And Hatch-Waxman know, theres a lot that happens before FDA can take action on an ANDA.
The US FDA’s Center for Devices and Radiological Health (CDRH) dropped a bevy of new digital health guidances and reports today and yesterday, providing some long-awaited clarity and peeks into the agency’s future plans. . The push started yesterday with the 31-page key findings report from the FDA Pre-Certification Program pilot.
Schwartz — FDA recently published a Federal Register (FR) Notice [ Docket No. FDA-2024-N-3945 ] announcing the publication of a draft strategy document, for public comment, outlining specific actions FDA plans to take to facilitate the use of innovative manufacturing technologies.
FDA advisors will scrutinise three cancer immunotherapies granted conditional approvals at a three-day meeting this week, to see if they should stay on the market. . Briefing documents published by the FDA ahead of the meeting suggest that discussion will focus on ongoing trials that may serve as alternative confirmatory studies.
Gibbs & Ana Loloei & Véronique Li, Senior Medical Device Regulation Expert — FDA has long touted the use of real-world evidence ( RWE ). FDA recognizes the potential of RWE to support regulatory submissions of medical devices and to inform benefit-risk analysis of such products, while assuring patients have timely access to devices.
On February 7, at a town hall organised to discuss clinical trial designs for gene therapies, FDA experts pushed pharma players to look for ways to establish clinical effectiveness despite the challenges in recruiting patients with rare diseases.
Prescribing Red Flags The government alleged that from at least 2017 to April 2021 Defendants knowingly filled controlled substance prescriptions “that raised obvious ’red flags’ of potential abuse or diversion.” If the pharmacist can resolve it, they must make a record of the resolution. Complaint ¶ 55.
Mullen — With comments due on the proposed LDT rule last week, FDA wasted no time updating the Unified Agenda to indicate that a final rule may be published in April ( here ). Under the Administrative Procedure Act, FDA is obligated to address major substantive issues when – and not so much if – it publishes a final rule.
Baumhardt, Senior Medical Device Regulation Expert & Philip Won — The Center for Devices and Radiological Health’s (CDRH) Standards and Conformity Assessment Program (S-CAP) encourages medical device sponsors to use FDA-recognized voluntary consensus standards in their product submissions.
Tobolowsky — Much has changed since the long-gone days of 2017. The Washington Nationals won the World Series, Presidential administrations have come and gone, and FDA has added new meeting types and formats to its menu. And so, FDA has issued a new draft guidance to bring everyone up to speed on formal meetings under PDUFA.
Lenz, Principal Medical Device Regulation Expert — FDA began the Software Precertification (Pre-Cert) pilot program in 2017 to evaluate an alternative approach to regulation of software as a medical device (SaMD) over the total product lifecycle (TPLC). As part of the pilot program, FDA included a Test Plan. By Adrienne R.
Although Ozempic has been approved by the Food and Drug Administration (FDA) since 2017, its status as a household name is relatively recent. Weight loss is an off-label , non-FDA-approved use for Ozempic. The FDA has urged consumers to use caution when taking medication from these compound pharmacies.
In the US, botanical dietary supplements can be sold without US Food and Drug Administration (FDA) approval, 1 prompting many vendors to promote the use of botanical products as dietary supplements , 2 rather than pursue a path of regulatory approval. 3 Is two too few? 3 Is two too few?
The first draft for comments from industry stakeholders was published in 2017 and generated 6000 comments. 12 of February 2020 again received 2,000 comments from public consultation.
Bob also initiated an agreement under which ISPE’s Guidance Documents are made available to PIC/S and WHO inspectors, which is still in place today. Phillips Professional Achievement Award in 2017 , an award that honors an ISPE Member’s significant lifetime contribution to the industry, not just ISPE.
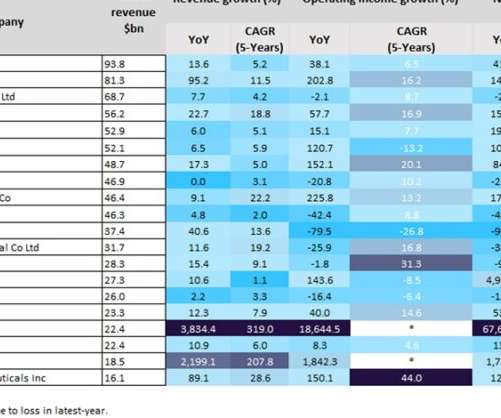
Bristol Myers Squibb (BMS) documented a 9.1% However, the FDA's approval of Lumakras for metastatic non-small cell lung cancer in May last year has a global analyst consensus sales forecast of $336m for this year, according to GlobalData’s Drugs Database. Amgen reported only a marginal 2.2% YoY revenue growth to $26.0bn last year.
These teams will include experts in regulatory, GMP compliance and quality systems, often including ex-FDA and MHRA inspectors and industry experts. She joined NSF in 2017. Preparing for acquisition means preparing for this due diligence investigation to maximise your value and attractiveness.
In 2020, the Federal Trade Commission issued a report regarding settlements reached between brand and generic manufacturers in FY 2017 and noted that “for the first time since [FY] 2004, no settlement agreement in [FY] 2017 contains a no-[authorized generic] AG commitment.”. Gilenya is an oral medication for multiple sclerosis.
The PD-1 inhibitor received an accelerated approval for this indication back in May 2017. Earlier in March, the FDA shared a draft guidance on how to run clinical trials for the accelerated approval of cancer drugs. However, not all accelerated approvals for Keytruda have been successfully converted into full approvals.
Of all products approved by the European Medicines Association (EMA) between 2017 and 2020, 68% are currently available in England, a figure that stands at 54% in Scotland. They show that England was sixth and Scotland tenth out of 13 countries when ranked according to the percentage of new medicines made available to patients.
This track will delve into five important topics where you will hear from speakers who will help answer your questions and provide you with the necessary information to make confident decisions regarding any updated changes to your facility, equipment, procedures, and documents.
156, as added by the 1984 Hatch-Waxman Amendments, for certain FDA-regulated products. The use of a surrogate endpoint can considerably shorten the time required prior to receiving FDA approval. If the confirmatory trial shows that the drug actually provides a clinical benefit, then the FDA grants traditional approval for the drug.
Improvements have been observed by various regulatory agencies in Latin America for the acceptance and implementation of international standards—for instance, the ICH Common Technical Document (CTD) format. Additional required information beyond ICH guidelines, or non-value-added documents (e.g., 6, and 2.3.S.7 7 are required.
The Advancing Pharmaceutical Quality, Quality Management Maturity Program includes five guidance documents: Corrective Action and Preventive Action (CAPA) : ICH Q10 demonstrates defined requirements for a robust corrective action and preventive action system throughout the product lifecycle. Problem Identification. Root Cause Identification.
We’re trying to register lifesaving therapies, but we have to collect the data manually, and often the FDA and EMA inspectors have to visit trial sites to validate the data. He calls out a particular opportunity in the United Kingdom, where COVID-19 and other factors have reduced patient access to clinical trials by 44% since 2017.
In addition, the OPEN Act, introduced in 2017, can extend the exclusivity period of a previously approved drug by another six months. Once the designation request has been submitted to the FDA, it is reviewed by the OOPD , which determines if all criteria for ODD approval have been met.
Dupixent (dupilumab) is a brand-name monoclonal antibody drug that is approved by the Food and Drug Administration (FDA) to treat a variety of conditions. Its first approval was in 2017 for atopic dermatitis (eczema). Most only issue coverage for treatments approved by the FDA.
In 2017 Oximio began providing dedicated local clinical trial support from its depot in Tbilisi as well as its courier fleet. Global inflation and the increased cost of petrol are also influencing the cost of multinational airfreight, making shipments from the EU and US more expensive than a distribution from a local depot. Expert partners.
The only FDA approved treatment for post-partum depression is a progesterone analogue that must be delivered over a 60-hour IV infusion and appears mildly efficacious. There have been a couple of small case series document LSD use in pregnancy and normal birth outcomes, although these are not high quality or conclusive by any means [23].
In Sep 2022, Konica’s Chiropractic Straight Arm (CSA) system received the US FDA’s approval as an X-ray technology allowing diagnosis through visualization of the anatomy. In Jul 2022, DiaSorin’s Liaison MeMed BV test received the US FDA’s approval for its commercial launch across the US.
Although the pharmaceutical industry has consistently improved manufacturing processes 3 in compliance with good manufacturing practices, 4 there are documented deviations from good practices 5 including the continued falsification of medicines. Published 19 January 2017. 30 June 2016. link] 6 BBC News. link] 7 He, J., Cai, and X.
Also, determining whether an intrusion occurred and documenting that intrusion adds to the challenges of performing aseptic technique properly (and perhaps adds to the subjectivity of the current control strategies). These media fill process simulations also serve to evaluate the aseptic technique used during operator interventions.
The FDA, PIC/S, and WHO have all emphasized the importance and benefits of data flows in their guidance on data integrity. North Bethesda, MD: International Society for Pharmaceutical Engineering, 2017. 3 , 4 RAMI integrates all assets, including physical items, software, administrative shell, documents, and personnel.
5 It includes cleaning validation master plan, product risk assessment, utility and equipment readiness, analytical method readiness, sampling site selection/grouping, standard operating procedures, validation protocols, execution of validation protocols, personnel training, and the validation documentation package. 11 (2017): 36–46.
The setpoint for “proper” air velocity in cleanroom systems is documented in standards and regulations as 0.45 9 In 2015, the FDA published a guidance manual 10 that provided questions for inspections, including: Is the air flow in critical areas unidirectional when delivered to the point of use? 2 (2017): 44–46. m/s up to 0.54
For changes requiring approval before implementation, once the necessary information has been submitted across regions, each health authority must review the data package and documentation based on local requirements. laboratory notebooks, batch records, and technical reports) to submission documents (e.g.,
Mullen — On December 9, 2022, FDA issued a draft guidance document on the Voluntary Malfunction Summary Reporting (VMSR) Program for medical devices. The purpose of this program is to reduce the volume of reports that a manufacturer needs to submit to FDA and to “make malfunction event trends more readily apparent.”.
When FDA published its draft guidance on payor communications in 2017, it seemed revolutionary – introducing the concept of pre-approval information exchange for wholly unapproved products. FDA had put industry in a quandary.
It’s also approved by the Food and Drug Administration (FDA) to treat schizophrenia. Adults, adolescents, and children 10 years and older Adults, adolescents, and children 7 years and older Latuda vs. lithium: Conditions treated Through rigorous clinical trials and documentation, the FDA approves medications for very specific uses.
Livornese — Last month, Congress took a big step towards improving clinical trial diversity by requiring sponsors of most drug and device clinical studies to submit a diversity action plan when they submit key trial documents to the Food and Drug Administration (FDA). This guidance was finalized in 2020. by the end of 2023).
I’m excited to report that since the initial publication of this article in 2017, various individuals and companies have created more accessible, direct-to-consumer devices to provide red light therapy; most of them use LEDs instead of lasers. This Class II medical device is FDA-cleared and meant to target all kinds of pain.
Cato — The long-running saga of FDA regulation of laboratory-developed tests (LDTs) has taken yet another new turn. The announcement in the Unified Agenda of a planned NPRM is the most concrete evidence that FDA is moving forward with this plan. Javitt & McKenzie E. So far, all of these efforts have been fruitless.
The past year has seen significant developments in the injectables landscape with the rapid introduction and development of vaccines in response to the pandemic, updates in regulations including the EU MDR and FDA guidance on bridging studies, and increasing industry acceptance of connectivity to aid the user experience. 08.30 – 12.30.
7 A critical feature of the ETP is representation from all relevant FDA quality assessment and inspection programs, from early engagement with stakeholders through application submission and assessment. 13 An FDA-authored paper indicating support for the implementation of CM using science- and risk-based approaches followed soon thereafter.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.














































Let's personalize your content