This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The US Food and Drug Administration (FDA) has sent a warning letter to KVK-Techs drug manufacturing facility following an inspection in April 2019. The document identified significant violations of current good manufacturing practice (cGMP) regulations for finished pharmaceuticals. Whats next for KVK-Tech?
As of last October, the agency had received more than 500 drug submissions with AI components going back to 2016, with a large number in the areas of oncology, neurology, and gastroenterology.
. | Ahead of an advisory committee meeting slated for Friday, the FDA unleashed damning briefing documents calling into question the effectiveness and safety of Intercept’s rare liver disease drug Ocaliva.
One medical device lawyer wrote that the document “violates” the 21st Century Cures Act passed by Congress in 2016 to exempt certain software products from regulatory review. But they are sharply divided on whether that’s a much-needed change, or a dramatic overreach by regulators.
Lewis, Senior Regulatory Device & Biologics Expert — FDA recently published the final guidance document “ Comparability Protocols for Postapproval Changes to the Chemistry, Manufacturing, and Controls Information in an NDA, ANDA, or BLA.” By Holly N. Brevig, Senior Regulatory Device and Biologics Exper & Richard A.
FDA advisors will scrutinise three cancer immunotherapies granted conditional approvals at a three-day meeting this week, to see if they should stay on the market. . Briefing documents published by the FDA ahead of the meeting suggest that discussion will focus on ongoing trials that may serve as alternative confirmatory studies.
It’s from 2016 about the future of antibiotics and resistance, but it remains highly relevant today and people consistently give positive feedback about it. Memorizing 45+ page document is certainly not a reasonable expectation, but one can certainly walk away with an awareness of general concepts and themes which are relevant.
The primary vehicle for FDA to request cybersecurity information in premarket submissions has been guidance documents. The primary vehicle for FDA to request cybersecurity information in premarket submissions has been guidance documents. It applies whether the software is the entire device (i.e.,
Users can also use the built-in health records feature on the Health app to download current medications, said Ricky Bloomfield, a former director of mobile strategy at Duke University who joined Apple as clinical and health informatics lead in 2016 in a tweet.
FDA , Petitioners, a liquid nicotine manufacturer, sued FDA arguing that the Agency was arbitrary and capricious in rejecting the Petitioner’s Premarket Tobacco Application (“PMTA”) in violation of the Administrative Procedure Act (“APA”). Youth behavioral data was not specifically required but FDA encouraged such information.
Mullen — More than five years after FDA first announced its plan to harmonize 21 CFR Part 820 with ISO 13485, on February 2, 2024, FDA finally issued the Quality Management System Regulation (QMSR) Final Rule. FDA further retained some definitions in the QSMR. The new § 820.10 Revised § 820.3
Mullen — On December 9, 2022, FDA issued a draft guidance document on the Voluntary Malfunction Summary Reporting (VMSR) Program for medical devices. The purpose of this program is to reduce the volume of reports that a manufacturer needs to submit to FDA and to “make malfunction event trends more readily apparent.”.
Shapiro — More than a decade ago, FDA began systematically to incorporate review of human factors (HF) design validation within 510(k) reviews. Now FDA has issued a draft guidance , Content of Human Factors Information in Medical Device Marketing Submissions (Dec. FDA did not require HF data as a basis for clearance of the predicate.
In the US, botanical dietary supplements can be sold without US Food and Drug Administration (FDA) approval, 1 prompting many vendors to promote the use of botanical products as dietary supplements , 2 rather than pursue a path of regulatory approval. 3 Is two too few? 3 Is two too few?
By Riëtte van Laack — On September 28, 2022, FDA announced the availability of the proposed rule for the implied nutrient content claim “healthy.” The term healthy, as an implied nutrient claim, was first defined by FDA in 1994. FDA also announced it would be re-evaluating the regulatory criteria for use of the “healthy” claim.
Food and Drug Administration ( FDA ) as a weight loss medication. As of now, phentermine is not FDA-approved for ADHD, and the evidence of its effectiveness is scarce. Phentermine is an old psychotropic medication that is indicated by the FDA for the short-term treatment of weight loss,” she says. “It
Javitt — On September 28, 2022, the FDA issued the long anticipated final Clinical Decision Support Software Guidance (CDS Guidance), which replaces the revised draft guidance document from 2019. FDA interprets the term “pattern” to mean “multiple, sequential, or repeated measurements of a signal or from a signal acquisition system.”
We now see that the proposed rule to “harmonize and modernize” the QSR with ISO13485:2016, creating the new QMSR, is on the Spring 2023 Unified Agenda (see here ). The final rule’s publication is a “high priority”, according to Dr. Once the final rule is published there will still be significant work to be done by FDA.
Livornese — FDA recently published a Draft Guidance entitled “Charging for Investigational Drugs under an Investigational New Drug Application: Questions and Answers” (the Draft Guidance). This Draft Guidance, when finalized, will replace the Final Guidance issued just six years ago (the 2016 Guidance). Changes from the 2016 Guidance.
156, as added by the 1984 Hatch-Waxman Amendments, for certain FDA-regulated products. The use of a surrogate endpoint can considerably shorten the time required prior to receiving FDA approval. If the confirmatory trial shows that the drug actually provides a clinical benefit, then the FDA grants traditional approval for the drug.
The paper, by a team from Imperial College London, concluded that the FDA gave the go ahead to 95% of the 89 products approved between 2010 and 2019 before EMA, with the Europeans trailing the Americans by a median of 241 days.
CDFA has established a web page with multiple resources for producers and distributors, including links to the regulations and guidance documents. Voters in Massachusetts approved a ballot initiative (Question 3) in 2016 that imposed requirements similar to those imposed under Proposition 12.
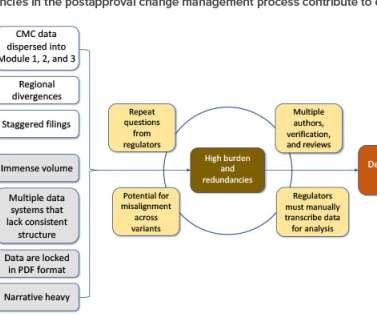
For changes requiring approval before implementation, once the necessary information has been submitted across regions, each health authority must review the data package and documentation based on local requirements. laboratory notebooks, batch records, and technical reports) to submission documents (e.g.,
The final presentation was Regulatory Considerations and Challenges in Development of Cell Therapy Products – CMC Perspective delivered by Dr. Melanie Eacho, Cell Therapies Branch Chief within the Office of Tissues and Advanced Therapies (OTAT) , US FDA/CBER. CDER/OPQ/OQS/FDA. CDER/FDA. Christian Woelbeling. Scientific Advisor.
The first well-documented case of multiple myeloma was reported in 1844 by renowned British surgeon Samuel Solly. 2001– FDA green lights revolutionary treatments. Just over a decade after it was developed by biochemist Nicholas Lyndon, Imatinib received US Food and Drug Administration (FDA) approval in 2001.
Improvements have been observed by various regulatory agencies in Latin America for the acceptance and implementation of international standards—for instance, the ICH Common Technical Document (CTD) format. Additional required information beyond ICH guidelines, or non-value-added documents (e.g., 6, and 2.3.S.7 7 are required.
Food and Drug Administration (FDA) certifies the Red Dye 40 as safe for human consumption. Apart from Red Dye 40, there are eight other certified color additives approved for use in food by the FDA: Blue No. The FDA has denied any link between the two for years. The controversy surrounding Red Dye 40 The U.S. 1, Blue No.
It’s also important to know that while ACV is often sold as a nutritional supplement in many forms, including liquid, tablet, and capsule, it’s not regulated by the FDA. A 2016 study found the combination of ACV and the blood pressure medication Procardia XL to lower blood pressure in rats better than ACV alone.
China still hasn’t approved western mRNA vaccines, despite submission, and the US FDA rejects Chinese-only trial data for cancer medicines. For pharma, the impact in critical areas such as the performance of newly marketed innovation has already been documented.
For example, an assessment for a facility primarily focused on US and EU markets would only consider GMPs and regulations from EudraLex and the FDA. FDA CFR Title 21 Parts 211, 600, and 1271; 8. , FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing–Current Good Manufacturing Practice, 11.
The only FDA approved treatment for post-partum depression is a progesterone analogue that must be delivered over a 60-hour IV infusion and appears mildly efficacious. There have been a couple of small case series document LSD use in pregnancy and normal birth outcomes, although these are not high quality or conclusive by any means [23].
Although the pharmaceutical industry has consistently improved manufacturing processes 3 in compliance with good manufacturing practices, 4 there are documented deviations from good practices 5 including the continued falsification of medicines. 30 June 2016. 3 (2016): 76–8. link] 6 BBC News. Published 19 January 2017.
12 April 2016. FDA Official Warns Manufacturers of Common Problems Found in Aseptic Operations.” www.raps.org/news-and-articles/news-articles/2023/3/fda-official-warns-manufacturers-of-common-problem 25 US Food and Drug Administration Center for Drug Evaluation and Research. October 2022. Madhyastha. Science 372, no. 8 Riley, S.
However, when the US Food and Drug Administration (FDA) introduced “Pharmaceutical cGMPs for the 21st Century — A Risk-Based Approach” 5 in September 2004, the paradigm shifted and these questions were asked: What is the probability that changing the airflow in a given clean area will affect the process or product in that area? It has 3.5
This article of mine discusses 19 thyroid-toxic products that have been banned by the FDA. Feel free to use my own health timeline as a guideline to document your health journey. Published 2016 Jul 19. Published 2016 May 24. 2016 Sep;124(9):1479-86. 2016 Sep;124(9):1479-86. Front Endocrinol (Lausanne).
Livornese — Last month, Congress took a big step towards improving clinical trial diversity by requiring sponsors of most drug and device clinical studies to submit a diversity action plan when they submit key trial documents to the Food and Drug Administration (FDA). This guidance was finalized in 2020. by the end of 2023).
Cato — The long-running saga of FDA regulation of laboratory-developed tests (LDTs) has taken yet another new turn. The announcement in the Unified Agenda of a planned NPRM is the most concrete evidence that FDA is moving forward with this plan. Javitt & McKenzie E. So far, all of these efforts have been fruitless.
7 A critical feature of the ETP is representation from all relevant FDA quality assessment and inspection programs, from early engagement with stakeholders through application submission and assessment. 13 An FDA-authored paper indicating support for the implementation of CM using science- and risk-based approaches followed soon thereafter.
In December 2021, the US FDA issued a cross-center draft guidance with recommendations on the use of digital health technology tools (DHTTs) to acquire data remotely from participants in clinical investigations for medical products. Table 3: Example classification of DHTs. .
In December 2021, the US FDA issued a cross-center draft guidance with recommendations on the use of digital health technology tools (DHTTs) to acquire data remotely from participants in clinical investigations for medical products. Table 3: Example classification of DHTs. .
The writing of requirements, design documentation, and test scripts, and the configuration of hardware and software, can be conducted from home. During the COVID-19 pandemic, the US FDA issued guidelines for remote evaluations of drug manufacturing and bioresearch facilities. Harlow, Essex, England: Pearson Education Limited, 2016.
A 2015 case report documented the thyroid labs of a man who had recurrent hyperthyroid episodes after three of his wife’s pregnancies! January, 2016. Like women, men can develop hormonal changes after the birth of a child, which can be linked to postpartum depression. [14] His wife had Hashimoto’s.) Chem Res Toxicol.
As of 2022, there were 15 drugs (see Table 1) manufactured using CM elements that have received FDA approval, with GSK, Pfizer, and Vertex owning approximately 60% of the market share, followed by Janssen/J&J with about 13%. Table 1: List of FDA-approved commercial products using CM elements.
In 1998, the Food and Drug Administration (FDA) defined an adaptogen as a new kind of metabolic regulator that has been proven to help in environmental adaptation and to prevent external harms. Published 2016 May 12. doi:10.1155/2016/1360386 [25] He JY, Ma N, Zhu S, Komatsu K, Li ZY, Fu WM. Published 2016 Jul 12.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.













































Let's personalize your content