This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Karst The FDA Reduction-in-Force (Termination)or RIF(T)announced last week has resulted in countless stories in the press and on personal LinkedIn accounts from those RIFd. As folks steeped in the world of generic drugs And Hatch-Waxman know, theres a lot that happens before FDA can take action on an ANDA. 314.150(c).
They were approved under the FDA accelerated pathway in 2009 and 2014, respectively, for treating a rare form of blood cancer. The The company’s final study plan was submitted to the FDA in 2022 and is expected to be completed by 2030 , according to FDA briefing documents published earlier this week.
Mullen — With comments due on the proposed LDT rule last week, FDA wasted no time updating the Unified Agenda to indicate that a final rule may be published in April ( here ). Under the Administrative Procedure Act, FDA is obligated to address major substantive issues when – and not so much if – it publishes a final rule.
In May of 2014 and February of 2019 , the FDA released final Guidance for Industry outlining the Agency’s policies and procedures regarding expedited development and review programs for new drugs and biologics intended to treat serious or life-threatening conditions. Image courtesy of qimono at pixabay.com.
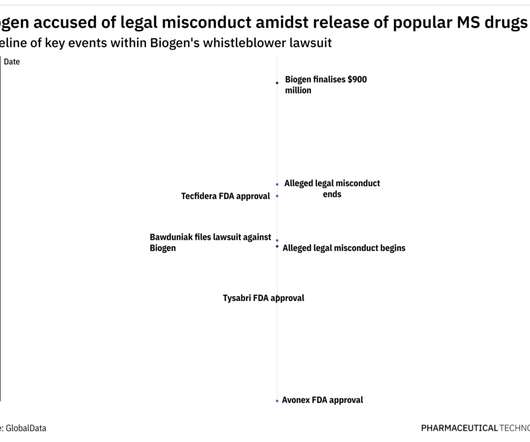
Mr. Bawduniak alleged that, between 2009 and 2014, Biogen paid illegal kickbacks to its largest prescribers to induce them to prescribe the company’s multiple sclerosis drugs, Avonex, Tysabri and Tecfidera, and discourage them from prescribing newer competitor products. This lawsuit was brought to the U.S. See United States ex rel.
In the US, botanical dietary supplements can be sold without US Food and Drug Administration (FDA) approval, 1 prompting many vendors to promote the use of botanical products as dietary supplements , 2 rather than pursue a path of regulatory approval. 3 Is two too few? 3 Is two too few?
The agreement outlined that Par would keep its generic equivalent off the market for as many as two years, delaying the generic launch till September 30, 2014. In September, Novartis sent the FDA a petition, after a similar one was rejected in April 2021, asking for Entresto’s patent protection to be extended to February 2024.
Broadening uses and secured full approvals Since its first approval in September 2014, Keytruda has achieved blockbuster status on the market. Earlier in March, the FDA shared a draft guidance on how to run clinical trials for the accelerated approval of cancer drugs.
Over the next year, IMPACT authored the following documents for the client: Pre-NDA meeting package. Working with the company’s electronic publishing vendor to ensure that fully-compiled, submission-ready documents were produced. 5 clinical study reports. and 2.7.4), and the Clinical Overview (Module 2.5).
Over the next year, IMPACT authored the following documents for the client: Pre-NDA meeting package. Working with the company’s electronic publishing vendor to ensure that fully-compiled, submission-ready documents were produced. 5 clinical study reports. and 2.7.4), and the Clinical Overview (Module 2.5).
A review of FDA’s Postmarketing Requirements and Commitments database reveals that one of the most common reasons FDA requires postmarketing studies is to assess the impact of a drug on maternal and fetal outcomes when taken by pregnant women. For post-approval pregnancy studies, it most certainly does.
156, as added by the 1984 Hatch-Waxman Amendments, for certain FDA-regulated products. The use of a surrogate endpoint can considerably shorten the time required prior to receiving FDA approval. If the confirmatory trial shows that the drug actually provides a clinical benefit, then the FDA grants traditional approval for the drug.
The case revolves around allegations that Novartis made an agreement in 2011 with Par Pharmaceutical when Exforge (valsartan/amlodipine) was nearing the end of its patent life that was designed to keep Par’s generic version of the drug off the market until 30 September, 2014.
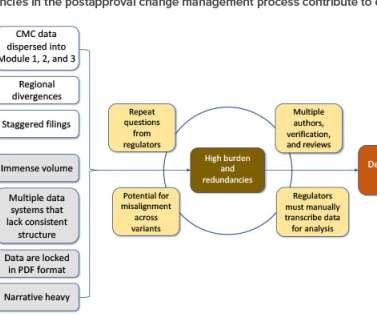
For changes requiring approval before implementation, once the necessary information has been submitted across regions, each health authority must review the data package and documentation based on local requirements. laboratory notebooks, batch records, and technical reports) to submission documents (e.g.,
It is estimated that only one in 10 drugs that enter Phase I trials are subsequently licensed by the US Food and Drug Administration (FDA). 5 When these medicines are prescribed to patients, we can use feedback from the real-world data that documents clinical outcomes, efficacy measures, patient-reported outcomes, and adverse events.
Between 2009 and 2014, the time period of Biogen’s alleged misconduct, the standard of care for MS involved the use of Biogen’s immunomodulatory drugs such as Avonex (Interferon beta-1a), Tecfidera (dimethyl fumarate), and Tysabri (natalizumab). 4 integrins, which play a key role in MS pathology.
Hosted software to publish regulatory submissions in the electronic common technical document (eCTD) format was deployed. The group successfully submitted a “pilot” eCTD submission to the Food and Drug Administration (FDA) and gained FDA approval to transmit submissions through the Electronic Submissions Gateway (ESG).
Additionally, a 2014 review of studies points to evidence that people with NAFLD are more likely to be deficient in vitamin C, leaving them more prone to inflammation and oxidative stress. Food & Drug Administration (FDA) like prescription drugs. It’s important to remember that herbal supplements aren’t regulated by the U.S.
Concept Paper: M4Q(R2) Common Technical Document on Quality Guideline.” As part of the US Food and Drug Administration (FDA) QbD pilot 4 in 2005–2006, the need to convey how the control strategy is linked to the target product profile (TPP) and quality target product profile (QTPP) was discussed. The authors propose using Module 2.3
Concept Paper: M4Q(R2) Common Technical Document on Quality Guideline.” As part of the US Food and Drug Administration (FDA) QbD pilot 4 in 2005–2006, the need to convey how the control strategy is linked to the target product profile (TPP) and quality target product profile (QTPP) was discussed. The authors propose using Module 2.3
For example, an assessment for a facility primarily focused on US and EU markets would only consider GMPs and regulations from EudraLex and the FDA. FDA CFR Title 21 Parts 211, 600, and 1271; 8. , FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing–Current Good Manufacturing Practice, 11.
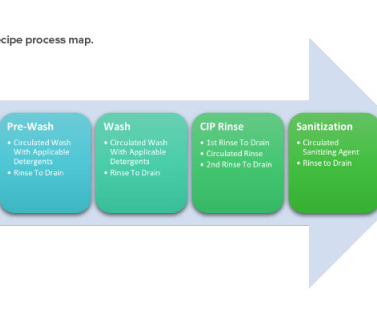
August 2014. 5 It includes cleaning validation master plan, product risk assessment, utility and equipment readiness, analytical method readiness, sampling site selection/grouping, standard operating procedures, validation protocols, execution of validation protocols, personnel training, and the validation documentation package.
10] I initially wrote a guide on LLLT for my clients in my Hashimoto’s Self-Management Program in 2014, before the follow-up study was done, and was concerned that the effects wouldn’t last. All Lumebox products are FDA-registered, follow the Good Manufacturing Practice (GMP) system, and every batch is third-party tested.
FDA Official Warns Manufacturers of Common Problems Found in Aseptic Operations.” www.raps.org/news-and-articles/news-articles/2023/3/fda-official-warns-manufacturers-of-common-problem 25 US Food and Drug Administration Center for Drug Evaluation and Research. September 2014. September 2022. 7 March 2023. Regulatory Focus.
USA The FDA established the Emerging Technology Program (ETP) in 2014 and has actively promoted the program 8. In his keynote presentation at the 2022 ISPE Annual Meeting & Expo in Orlando, FL, Dr. Michael Kopcha, Director of the Office of Pharmaceutical Quality, emphasized the FDA’s commitment to promoting advanced manufacturing.
Back in 2014, I found that my Hashimoto’s flared up after using a particular lip gloss; once tested, it came back as containing arsenic! . This article of mine discusses 19 thyroid-toxic products that have been banned by the FDA. Feel free to use my own health timeline as a guideline to document your health journey.
Surgeon General Luther Terry’s landmark Report, “ Smoking and Health ,” which definitively documented the causal link between smoking, disease, and death. Tobacco control policies implemented between 1964 and 2014 saved 8 million lives in the United States. In
In 2014, CDER established the Emerging Technology Program (ETP) within its Office of Pharmaceutical Quality to better facilitate the adoption of emerging technologies such as CM. 13 An FDA-authored paper indicating support for the implementation of CM using science- and risk-based approaches followed soon thereafter.
Livornese — Last month, Congress took a big step towards improving clinical trial diversity by requiring sponsors of most drug and device clinical studies to submit a diversity action plan when they submit key trial documents to the Food and Drug Administration (FDA). This guidance was finalized in 2020. by the end of 2023).
In December 2021, the US FDA issued a cross-center draft guidance with recommendations on the use of digital health technology tools (DHTTs) to acquire data remotely from participants in clinical investigations for medical products. Table 3: Example classification of DHTs. .
In December 2021, the US FDA issued a cross-center draft guidance with recommendations on the use of digital health technology tools (DHTTs) to acquire data remotely from participants in clinical investigations for medical products. Table 3: Example classification of DHTs. .
The writing of requirements, design documentation, and test scripts, and the configuration of hardware and software, can be conducted from home. During the COVID-19 pandemic, the US FDA issued guidelines for remote evaluations of drug manufacturing and bioresearch facilities. Los Angeles, CA, US: Sage Publications, 2014.
As of 2022, there were 15 drugs (see Table 1) manufactured using CM elements that have received FDA approval, with GSK, Pfizer, and Vertex owning approximately 60% of the market share, followed by Janssen/J&J with about 13%. Table 1: List of FDA-approved commercial products using CM elements.
In 1998, the Food and Drug Administration (FDA) defined an adaptogen as a new kind of metabolic regulator that has been proven to help in environmental adaptation and to prevent external harms. 2014 Apr;5(4):635-44. [12] Published 2014 Dec 18. Published 2014 Aug 15 [35] Gao Y, Wei Y, Wang Y, Gao F, Chen Z. Food Funct.
FDAs withdrawal authority when a confirmatory trial is not conducted with due diligence was expanded to include that FDA could specify the conditions for a postapproval study. Considering FDAs new authority to specify the conditions for such confirmatory trials (e.g.,
1 Following the current trend, there has been a significant rise in the number of documents mentioning both artificial intelligence (AI) and rare diseases. For documents published in 2014 this number was just six; in 2024 it was 157 ( Figure 2 ). 1 Figure 1 : Documents by year. 1 Figure 1 : Documents by year.
Change control process requirements vary between companies and regions, but here is a list of the most common ones to give you a general idea: EU 1252/2014 This is a guideline for companies operating in the European Union and dealing with active substances. FDA 21 CFR Part 211 This regulation governs the practices of U.S.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.

























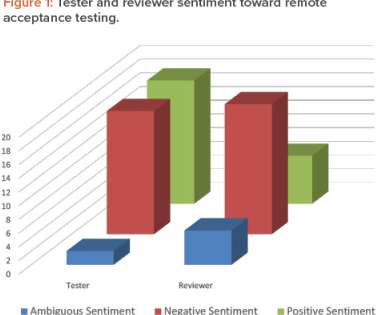











Let's personalize your content