This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
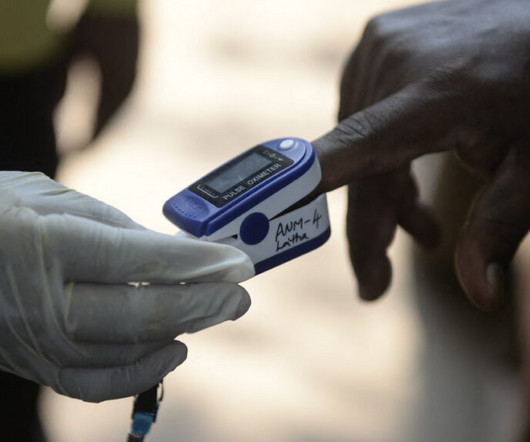
Work by device manufacturers to improve the performance of pulse oximeters on people with darker skin has progressed little since the Food and Drug Administration asked manufacturers in 2013 to voluntarily test the devices on more diverse skin tones, according to a study published Monday in JAMA.
Less than half of the cancer therapies given accelerated approval by the FDA in 2013 to 2017 showed a clinical benefit in a confirmatory trial within the next five years
One hallmark of the FDA's accelerated approval pathway is the requirement that medicines prove their benefits in confirmatory trials. Many cancer meds approved under the FDA's accelerated approval pathway from 2013 to 2017 didn't improve overall survival after more than five years of follow-up research, the study found.
In late 2013, the FDA had conditionally approved the biopsies as a part of an early feasibility study, which it limited to six patients at the medical center who were getting DBS for treatment-resistant depression. The review, which lists consultations with 16 different FDA officials, was scathing.
In some cases, failure to show clinical benefit didn’t stop the FDA from converting accelerated approvals into full approvals, and the authors note the agency’s conversion decisions have increasingly been based on less stringent evidence of a drug’s benefits. In 2013, it took an average of 9.9
A federal law that went into effect in 2013 requires the FDA to identify ingredients needed to satisfy an unmet “clinical need” and to include those on a list for use by large compounding pharmacies. Continue to STAT+ to read the full story…
Founded in 2013, Pear was the most prominent company developing prescription digital therapeutics, or Food and Drug Administration-cleared software applications ordered by doctors to treat health conditions. A hearing to approve the sale will be held on May 22.
After more than six years of development, Angle has scored an FDA approval for Parsortix as a diagnostic for metastatic breast cancer – the first ever in the US for a device used to detect cancer by harvesting cells from a patient blood sample. The post Angle claims FDA okay for Parsortix liquid biopsy system appeared first on.
So judging the FDA’s accelerated approval program without assessing its full impact on overall survival presents a very slanted story. Overall survival is usually considered the gold standard in oncology because people with cancer generally want to take medications that can help them live longer.
The FDA has started a priority review of Chiesi ‘s velmanase alfa, an enzyme replacement therapy for lysosomal storage disease (LSD) alpha-mannosidosis, with a decision expected in the first half of 2023. The post FDA starts review of first drug for alpha-mannosidosis, from Chiesi appeared first on.
Hospitals ran short of essential treatment medications and were unable to source those drugs from manufacturers or from the outsourcing facilities that had been authorized by Congress in 2013 to “fill the gap” in such situations. Read the rest…
The two men worked together from 2013 until 2022, when Jonas left to start a biotech incubator with funding from investment giant CBC Group. The VC firm has hired Jeff Jonas, Sage’s former chief executive, and Al Robichaud, Sage’s former chief scientific officer, as partners, the team told STAT exclusively.
Why, then, are FDA inspections abroad so infrequent and disappointing? FDA Inspections Suspended: During the COVID pandemic, the Food and Drug Administration suspended its inspections at most foreign pharmaceutical facilities. Even under the best of conditions, the FDA never inspects all foreign pharmaceutical plants annually.
elaborated, noting that based on recall data collected by the US Food and Drug Administration (FDA) in over 100 products between 2000–2010, “fungi existed in 21 percent of products.” The authors also shared: “in the last three years [since 2023], the US FDA has recalled several drugs due to fungal contamination.” Ahmed et al.
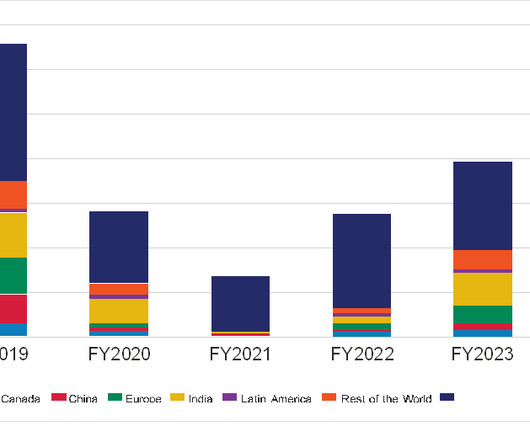
The report, Clinical Trials – The Importance of Diversity in Clinical Trials , examined data from clinical trials initiated between 1 January 2013 and 16 June 2022. Earlier this year the US Food and Drug Administration (FDA) published draft guidance to support companies in enrolling more ethnically diverse trial populations.
Safety concerns seem to have scuppered any hope of a near-term approval for FibroGen and AstraZeneca’s roxadustat for anaemia associated with chronic kidney disease (CKD) in the US, after FDA advisors voted comprehensively against the drug yesterday.
GSK licensed otilimab from German biotech MorphoSys in 2013 in a deal valued at up to €423 million, including around €23 million upfront. Daprodustat backed. There has been somewhat better news for GSK for another of its top pipeline prospects – renal anaemia drug daprodustat – although that also came with disappointment.
New Zealand medical tech startup Alimetry has claimed an FDA approval for a wearable device that can be used to diagnose gastric disorders non-invasively. Functional dyspepsia alone was estimated to cost the US upwards of $18 billion in healthcare expenses, according to figures published in 2013.
” Baynes joined Merck in 2013, a year before the company launched its cancer blockbuster Keytruda (pembrolizumab) which has since grown into a $17 billion-a-year product and racked up FDA approvals across more than 15 cancer types.
Sasinowski — On December 12, 2023, FDA announced the creation of a new advisory committee specifically for treatments for genetic metabolic diseases, the Genetic Metabolic Diseases Advisory Committee, or “GeMDAC.” Note that FDA is currently soliciting applications to staff this committee. Tobolowsky & James E. Raver & Frank J.
The application of Bayesian methodology has been recognised by the US Food and Drug Administration (FDA) as useful in early phase clinical trials involving paediatric populations. 2013 Oct; 48(10): 943–953. Published online 2013 Jul 2. FDA presentation. Regulatory and payer guidance. Pediatr Pulmonol. doi: 1002/ppul.22693.
Invokana was the first SGLT2 inhibitor to reach the market for diabetes after it was cleared by the FDA in 2013, and sales accelerated to a peak of nearly $1.4 billion in 2016 before the product was linked to an increased risk of lower limb amputation.
As industry is well aware, by the terms of the statute, the DSCSA’s interoperability provisions become effective ten years after its 2013 enactment, or on November 27, 2023. By Karla L. the ability to associate the saleable return product with the transaction information/statement with the particular product).
On October 17, 2022, FDA published the list of CDRH proposed guidances for FY 2023 (see here ). These are documents on the A-list, a list of prioritized documents that FDA intends to publish during FY2023. FDA published draft guidances for these topics in December 2021 ( here and here ).
The US Food and Drug Administration (FDA) has recently expressed concern to the US PTO about some types of innovator patent strategies that potentially delay generic entry into the market. FDA highlighted that over three quarters of new patents in the Orange Book between 2005 and 2015 were assigned to existing drugs. 23 April 2013.
18% of study participants were Black/African American in 2013, but that contracted to 14% in 2020 and then plunged to just 9% in 2021. For Hispanics, participation in trials increased from 7% in 2012 to a high of just under 10% in 2017 but fell back to levels of around 8% to 9% in the past two years.
This also occupies a large resource, given the US Food and Drug Administration (FDA) requirement for double plate checking using a second ‘independent’ person. The FDA is doing this through the publications of Guidance for Industry. In the recent FDA guidance on nanomedicine, the microbiological control aspects are featured.
Claud — FDA’s Office of Pharmaceutical Quality (OPQ) in the Center for Drug Evaluation and Research (CDER) is charged with assuring that drugs marketed in the U.S. While no office at FDA truly works in a vacuum, we can safely call OPQ the tip of FDA’s quality spear. In FY2021 and FY2022 combined, FDA added 77 companies.
Trintellix (vortioxetine) is a brand-name prescription medication that was approved by the Food and Drug Administration (FDA) in 2013 to treat major depressive disorder (MDD) in adults. It belongs to a class of drugs known as serotonin modulators and stimulators (SMS) that balance serotonin, a substance in the brain that affects mood.
Regulatory speakers were Lisa Hedman, Group Lead for the Division of Supply and Access to Medicines of the World Health Organization (WHO); and Joyce Cirunay, Director IV of the Center for Drug Regulation and Research at the Philippines FDA. Hustead Executive Director, Regulatory Affairs Merck & Co.,
Food and Drug Administration (FDA) used to rely heavily on a letter-based rating system for drug safety during pregnancy , with category A drugs being the safest to take, category X drugs being contraindicated because the risks outweigh the benefits, and category B, C, and D drugs varying in safety and adverse effects.
Food and Drug Administration (FDA) recently approved Zepbound , a new version of Mounjaro, for weight loss. It can help trigger a feeling of fullness after a meal and slow the digestion process. Tirzepatide is manufactured under the brand name Mounjaro for Type 2 diabetes treatment.
billion in annual earnings as a result of unmanaged mental illness, and depression alone cost the global market $1 trillion in lost productivity in 2013. This year, Akili received FDA clearance for their digital therapeutic for children with ADHD by forcing the brain to adapt with a cleverly designed video game that forces focus.
Vibegron (brand name: Gemtesa) is the latest drug used to treat overactive bladder that has been approved by the FDA in the USA. Mirabegron is reserved as an optional treatment in the symptoms of overactive bladder in patients whom (NICE, 2013): . NICE, (2013). In this post, I will summarise: . Propiverine hydrochloride.
we must prove the effectiveness of radiopharmaceuticals through clinical trials… This has been demonstrated recently with PLUVICTO… the first US Food and Drug Administration (FDA)-approved targeted radioligand therapy for eligible patients with prostate cancer” At this moment, there is no single treatment for cancer today.
AbbVie has filed with the FDA to extend the uses of its bipolar disorder therapy Vraylar to include adjunctive therapy for major depressive disorder, part of a plan to build sales of the drug to peak sales of $4 billion or more. Analysts at GlobalData are predicting peak sales of $383 million for the drug in 2029.
In 2013, the US Food and Drug Administration (FDA) approved Ravicti (glycerol phenylbutyrate), manufactured by Hyperion Therapeutics, which has since then been acquired by Horizon Pharma. Drugs for dialysis, amino acid supplements, and drugs which convert blood ammonia are marketed right now as the go-to urea cycle disorder treatments.
15, 2013 ). Researchers at a hospital in Honolulu (Kaiser Permanente Hawaii) tracked patients who had treatment for RDHD between 2013 and 2016. We try to be your Drug Watchdog, keeping an eye on Big Pharma, the generic drug industry and the FDA. Some people report insomnia and anxiety after an injection.
Food and Drug Administration (FDA). Functions and action mechanisms of flavonoids genistein and icariin in regulating bone remodeling , Journal of Cellular Physiology (2013). Consumption of flaxseed, a rich source of lignans, is associated with reduced breast cancer risk , Cancer Causes & Control (2013). Molecules (2019).
FDA has been implementing the UDI system with different compliance dates for different types of medical devices to ensure a smooth implementation. The compliance dates were first published in 2013, and subsequently updated in various guidance documents and regulations published by FDA. See 21 CFR 801.40(d).
.” KW-6356 is a follow-up to Kyowa Kirin’s first-generation adenosine A2 antagonist Nourianz/Nouriast (istradefylline), which finally claimed FDA approval in 2019 as an add-on to levodopa in Parkinson’s after more than two decades of R&D and has been available in Japan since 2013.
In fact, in late 2023, its manufacturer, Eli Lilly, released another brand form of tirzepatide, Zepbound , with Food and Drug Administration (FDA) approval for weight loss. Still, according to the FDA’s boxed warning , anyone with a history of MTC or a personal or family history of MEN2 should avoid it.
Palmer — Last week FDA published a long-awaited Draft Guidance for outsourcing facilities addressing the Prohibition on Wholesaling Under Section 503B of the Federal Food, Drug, and Cosmetic Act (Draft Guidance). By Kalie E. Richardson & Karla L. See 21 U.S.C. Section II at 2.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.

















































Let's personalize your content