This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Karst The FDA Reduction-in-Force (Termination)or RIF(T)announced last week has resulted in countless stories in the press and on personal LinkedIn accounts from those RIFd. As folks steeped in the world of generic drugs And Hatch-Waxman know, theres a lot that happens before FDA can take action on an ANDA.
Work by device manufacturers to improve the performance of pulse oximeters on people with darker skin has progressed little since the Food and Drug Administration asked manufacturers in 2013 to voluntarily test the devices on more diverse skin tones, according to a study published Monday in JAMA.
In study documents, Mount Sinai doctors said the biopsies result in “the same amount of tissue loss” and “in effect, the same level of risk” for patients as standard DBS, because they are removing tissue that would otherwise be cauterized.
Why, then, are FDA inspections abroad so infrequent and disappointing? FDA Inspections Suspended: During the COVID pandemic, the Food and Drug Administration suspended its inspections at most foreign pharmaceutical facilities. Even under the best of conditions, the FDA never inspects all foreign pharmaceutical plants annually.
On October 17, 2022, FDA published the list of CDRH proposed guidances for FY 2023 (see here ). These are documents on the A-list, a list of prioritized documents that FDA intends to publish during FY2023. FDA published draft guidances for these topics in December 2021 ( here and here ).
Safety concerns seem to have scuppered any hope of a near-term approval for FibroGen and AstraZeneca’s roxadustat for anaemia associated with chronic kidney disease (CKD) in the US, after FDA advisors voted comprehensively against the drug yesterday.
The application of Bayesian methodology has been recognised by the US Food and Drug Administration (FDA) as useful in early phase clinical trials involving paediatric populations. 2013 Oct; 48(10): 943–953. Published online 2013 Jul 2. FDA presentation. Regulatory and payer guidance. Pediatr Pulmonol. doi: 1002/ppul.22693.
He warned, “Implementation of a CCS will require more than writing documents.”. This also occupies a large resource, given the US Food and Drug Administration (FDA) requirement for double plate checking using a second ‘independent’ person. The FDA is doing this through the publications of Guidance for Industry.
The FDA granted full approval to Par’s generic on 28 March, 2013. According to court documents cited by Reuters, annual sales of Exforge in the US were over $400 million before generic versions reached the market.
In the United States, the Food and Drug Administrator (FDA) has so far published three draft guidance documents on patient-focused drug development, with a fourth expected in the coming months. EMA and FDA backing represents a huge step forward for patient centricity as a force for good in pharmaceutical development.
FDA has been implementing the UDI system with different compliance dates for different types of medical devices to ensure a smooth implementation. The compliance dates were first published in 2013, and subsequently updated in various guidance documents and regulations published by FDA. See 21 CFR 801.40(d).
Improvements have been observed by various regulatory agencies in Latin America for the acceptance and implementation of international standards—for instance, the ICH Common Technical Document (CTD) format. Additional required information beyond ICH guidelines, or non-value-added documents (e.g., 6, and 2.3.S.7 7 are required.
Food and Drug Administration (FDA) certifies the Red Dye 40 as safe for human consumption. Apart from Red Dye 40, there are eight other certified color additives approved for use in food by the FDA: Blue No. The FDA has denied any link between the two for years. The controversy surrounding Red Dye 40 The U.S. 1, Blue No.
In 2013, the IMPACT (now part of Syner-G BioPharma Group) management team decided that the company needed to build a Regulatory Operations department to support their existing and future client base. Hosted software to publish regulatory submissions in the electronic common technical document (eCTD) format was deployed.
Exploring the Drug Supply Chain Security Act (DSCSA) The DSCSA, signed into law in 2013, aims to heighten the security of the pharmaceutical supply chain by creating a framework for traceability and accountability. This record-keeping includes detailed documentation of the product’s history and movement through the supply chain.
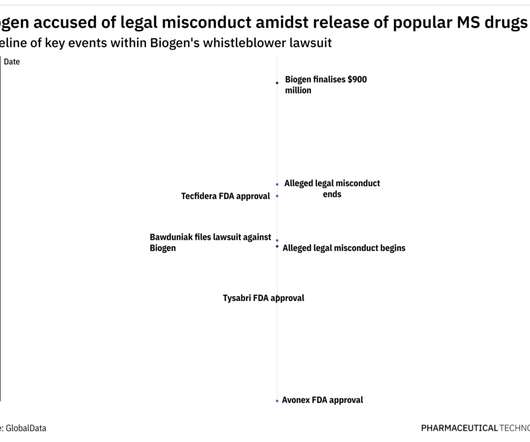
These, along with other similar claims, meant Biogen owed potential damages of $1,036,900,151 to the US and the various States, as per court documents. Tecfidera was approved in 2013 to treat MS, and in August 2020, Mylan launched its first FDA-approved dimethyl fumarate generic to the market.
Even worse, it showed flaws in our domestic system of drug regulation, notably the Drug Supply Chain Security Act of 2013 (DSCSA). To the extent that SafeChain sold any counterfeit medication, SafeChain was an unwitting participant and notified the FDA of customer complaints as required by law. Where’s the FDA?
The only FDA approved treatment for post-partum depression is a progesterone analogue that must be delivered over a 60-hour IV infusion and appears mildly efficacious. There have been a couple of small case series document LSD use in pregnancy and normal birth outcomes, although these are not high quality or conclusive by any means [23].
Although the pharmaceutical industry has consistently improved manufacturing processes 3 in compliance with good manufacturing practices, 4 there are documented deviations from good practices 5 including the continued falsification of medicines. 47 (18 November 2013): 12359–63. Angewandte Chemie International Edition in English 52, no.
Livornese — Last month, Congress took a big step towards improving clinical trial diversity by requiring sponsors of most drug and device clinical studies to submit a diversity action plan when they submit key trial documents to the Food and Drug Administration (FDA). This guidance was finalized in 2020. by the end of 2023).
This article of mine discusses 19 thyroid-toxic products that have been banned by the FDA. Feel free to use my own health timeline as a guideline to document your health journey. The Environmental Working Group (EWG) also has consumer resources that can help individuals source cleaner options. Published 2018 Jul 2. Int J Mol Sci.
In December 2021, the US FDA issued a cross-center draft guidance with recommendations on the use of digital health technology tools (DHTTs) to acquire data remotely from participants in clinical investigations for medical products. Table 3: Example classification of DHTs. .
In December 2021, the US FDA issued a cross-center draft guidance with recommendations on the use of digital health technology tools (DHTTs) to acquire data remotely from participants in clinical investigations for medical products. Table 3: Example classification of DHTs. .
Farquhar — A recent Warning Letter reflects an FDA citation of a company for refusing to permit FDA Investigators to take photographs during an inspection. FDA reports that it told the company that “failure to allow photography would be documented as a refusal,” and the company “acknowledged the refusal.” By Douglas B.
In 1998, the Food and Drug Administration (FDA) defined an adaptogen as a new kind of metabolic regulator that has been proven to help in environmental adaptation and to prevent external harms. doi:10.1155/2013/934183 [28] Siberian Ginseng. doi:10.1155/2013/514049; Cropley M, Banks AP, Boyle J. 2013;2013:934183. Anc Sci Life.
A few years ago, I had a few clients with documented elevations in prolactin, and I have seen that lowering prolactin levels can help with reducing thyroid antibodies. Studies have, however, documented that bromocriptine, a medication used to lower prolactin levels, can reduce flares in lupus, another autoimmune condition. Sex Med Rev.
To give examples, such a work package focuses on the identification of regulations (ICH Q2 R2, 3 Q8-Q14, 4-10 FDA PAT guidance, 11 EMA Annex 15 12 ), some of which may be mainly guidance documents with recommendations and flexibility when it comes to PAT.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.






























Let's personalize your content