This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
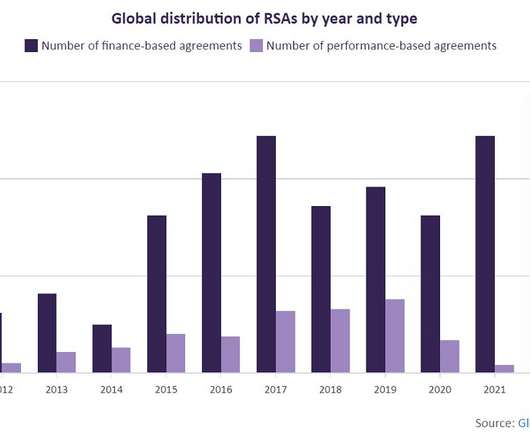
Data suggest confirmatory trials conducted for drugs granted accelerated approval by the FDA have taken over 3 years on average to complete since 2012.
The Food and Drug Administration’s rare pediatric disease priority review voucher program , which has been providing incentives for lifesaving innovations since 2012, is doomed to disappear unless Congress reauthorizes it before the end of September. I have data showing it works. None worked. Read the rest…
” That train of thought — calculating risks, eliminating threats — eventually led her to a job with the Food and Drug Administration in 2012 as a public health analyst. And I thought, doesn’t that mean nobody should really be sliding down poles?”
The US Food and Drug Administration (FDA) has approved GlaxoSmithKline Biologicals’ Boostrix vaccine for use in pregnant woman in their third trimester, to prevent against whooping cough (pertussis) in infants up to two months of age. Since 2012, the CDC has recommended the vaccine series during the third trimester of each pregnancy.
The traditional approvals follow initial accelerated approvals granted by the FDA in 2012 and the European Commission in 2014. It took many years, but Johnson & Johnson’s tuberculosis med Sirturo can finally claim full approvals in the U.S. and Europe following initial conditional nods. |
Under the agreement with the Swiss Confederation (Switzerland), the Swiss Agency for Therapeutic Products (Swissmedic) and the US Food and Drug Administration (FDA) will be able to utilise each other’s GMP inspections of pharmaceutical manufacturing facilities, avoiding the need for duplicate inspections.
In 2012, Ranbaxy paid a $350 million fine to U.S. As a result, the company has halted exports from the plant to the United States. At issue is a facility Sun bought eight years ago from Ranbaxy Laboratories, which was a poster child for lax manufacturing standards in India.
Designed in 2012 to encourage drug development for rare childhood diseases, the program provides vouchers to groups who get a drug in that area approved. The voucher is like a fastpass at Disneyland, allowing whoever has it to request the FDA review a drug within six months, vs. the usual 10. Read the rest…
The US Food and Drug Administration (FDA) created live biotherapeutic products (LBP) as a new category in the 2012 guidelines. EMA and FDA have given prime importance to the whole genome sequence characterisation of the strain in the final product dossier.
Gibbs & Ana Loloei & Véronique Li, Senior Medical Device Regulation Expert — FDA has long touted the use of real-world evidence ( RWE ). FDA recognizes the potential of RWE to support regulatory submissions of medical devices and to inform benefit-risk analysis of such products, while assuring patients have timely access to devices.
Who better than people living with a condition to inform drug companies, physicians, academics, and the FDA on what it is like to live with their condition, what symptoms most impact their lives, what goes into their decision about whether to participate in a clinical trial, and what kind of treatment effects would be most meaningful to them?
The US Food and Drug Administration (FDA) has approved GlaxoSmithKline ’s (GSK) Boostrix (tetanus toxoid, reduced diphtheria toxoid, and acellular pertussis vaccine adsorbed [Tdap]) for pregnant women during their third trimester to prevent pertussis (whooping cough) in newborn infants.
In the US, botanical dietary supplements can be sold without US Food and Drug Administration (FDA) approval, 1 prompting many vendors to promote the use of botanical products as dietary supplements , 2 rather than pursue a path of regulatory approval. 3 Is two too few? 3 Is two too few?
That has resulted in 26 partnerships, but the poster child of AbCellera’s portfolio is its alliance with Lilly for bamlanivimab (LY-CoV555), which was approved by the FDA and Canadian regulator for emergency use as treatment for mild-to-moderate COVID-19 patients last month.
Between 2012 and 2021, outsourcing propensity for FDA NDA injectables approvals averaged 40%, according to GlobalData’s Contract Injectable Packaging Trends…
Safety has been an issue throughout development and in 2012 the FDA put tanezumab on clinical hold because of a class-related issue with joint destruction, which was finally lifted in 2015. Stopping the drug after progression to more serious disease does not appear to be effective in preventing further damage to joints, the FDA added.
The FDA approved Truvada as the first PrEP treatment in July 2012, with Descovy receiving a nod for this use in October 2019. Truvada has since gone generic, with Teva Pharmaceuticals launching the first FDA-approved Truvada generic in October 2020. Gilead markets both Truvada and its successor Descovy.
The new EU approval comes after the FDA approved the expanded label last year, and provides the first treatment option that tackles the underlying cause of CF in patients aged six to 11 with these mutations. Together these drugs cover the CFTR mutations seen in around half of all CF patients.
Growth has been rapid since Europe’s first gene therapy approval in 2012, 2 and the first US Food and Drug Administration (FDA) approval of a gene therapy in 2017. 3 Any sector would struggle to keep up with expansion of this speed, but the inherent difficulty of working with and producing biologics adds another layer of complexity.
The FDA has concluded its safety review of Pfizer’s JAK inhibitor Xeljanz and Xeljanz XR, requiring revised warnings for the drugs as well as others in the class after finding evidence of elevated risks of serious heart-related events. The post FDA firms up JAK inhibitor warnings after Xeljanz review appeared first on.
Black/African American participation in trials has been declining over the past decade, says the report, falling from around 12% in 2012 to less than 10% in 2019 and 2020. For Hispanics, participation in trials increased from 7% in 2012 to a high of just under 10% in 2017 but fell back to levels of around 8% to 9% in the past two years.
Xeljanz has been a slow grower since its first approval for RA in 2012, but gradually grew into a blockbuster as clinicians gathered more experience with the drug and it picked up additional indications in psoriatic arthritis, ulcerative colitis and juvenile idiopathic arthritis.
Another 2012 article suggests that a formula containing shilajit and vitamin B complex may be effective against Alzheimer’s disease. In a 2012 animal study , researchers induced chronic fatigue syndrome (CFS) in rats for 21 days. Supplements are not regulated by the Food and Drug Administration (FDA), so contamination is possible.
Livornese — On February 6, 2024, FDA issued a draft guidance titled Notifying FDA of a Discontinuance or Interruption in Manufacturing of Finished Products or Active Pharmaceutical Ingredients Under Section 506C of the FD&C Act. By Véronique Li, Senior Medical Device Regulation Expert & Deborah L.
On November 22, 2022, the FDA approved CSL Behring’s Hemgenix (etranacogene dezaparvovec), the first gene therapy treatment for hemophilia B, with a staggering manufacturer price of $3.5 million, according to GlobalData’s Price Intelligence (POLI), making it the most expensive drug in the world.
Senate Committee on Health, Education, Labor and Pensions (“Senate HELP”) is scheduled to take up legislation that could significantly limit access to the courts and immunize critical FDA decisions from timely judicial review. Subsection 505(q) initially was added by Section 914 of the 2007 FDA Amendments Act (“FDAAA”), Pub.
Cato — On August 3rd, FDA issued 11 warning letters to foreign facilities registered as OTC drug manufacturers. For each of these facilities, FDA did not conduct an on-site inspection of the facility prior to issuing the warning letter. By McKenzie E.
Johnson & Johnson’s Darzalex Faspro has become the first product approved by the FDA to treat light chain (AL) amyloidosis, a rare and often fatal blood cell disorder.
Other drugs used to treat ATL include Kyowa Kirin’s amti-CCR4 antibody Poteligeo (mogamulizumab), cleared by the MHLW for CCR4-positive ATL in 2012, as well as Bristol-Myers Squibb/Celgene’s oral therapy Revlimid (lenalidomide), which got a green light there five years ago.
At the moment, the only FDA-approved treatments for ATTR cardiomyopathy are Pfizer’s Vyndaqel (tafamidis meglumine) and Vyndamax (tafamidis), which were given a green light by the agency two years ago. In ATTR amyloidosis, transthyretin (TTR) protein misfolds and accumulates as amyloid deposits throughout the body. billion by 2025.
Food and Drug Administration ( FDA ). The FDA explains that their regulations require a proposed expiration date for medications to be approved. The FDA agrees, urging people not to consume expired medicines and warning about the potency and safety risks. Adderall can expire, according to the U.S.
percent of recalls recorded by the US Food and Drug Administration (FDA) between 2012 and 2019. Burkholderia species are one of the primary causes of non-sterile pharmaceutical product recalls and accounted for 45.3
In 2012, the FDCA was modified to allow the submission of a De Novo request without the need for a prior 510(k), and set a target review time by FDA of 120 days. For example, FDA granted seven De Novos for COVID-19 related indications for use. It is important that FDA posts the decision summaries in a timely fashion.
district court of Massachusetts by former employee and whistleblower Michael Bawduniak in April 2012 as a qui tam action. Wasserstein — On September 26, 2022, Biogen Inc. agreed to pay $900 million to the United States and various states to settle a False Claims Act (FCA) lawsuit. This lawsuit was brought to the U.S. See United States ex rel.
This decision was granted a couple of weeks following approval by the US and Drug Administration (FDA). World Allergy Organization (WAO) White Book on Allergy 2011-2012: Executive Summary. What are the main advantages of EURneffy over similar treatments? Hand (N Y). 2007; 2(1):5-11. Derived from IQVIA Claims Data, 2023.
In March 2020, the FDA expanded the biosimilar category to include 90 additional molecules, meaning there are now more therapies that can serve as reference products for biosimilars. At the same time, the average cost of insulin for patients per year almost doubled from $2,864 to $5,705 between 2012 and 2016.
In 1990, the US Food and Drug Administration (FDA) requested Astra to change Losec’s brand name to Prilosec, to avoid confusion with Sanofi-Aventis’ diuretic, Lasix (furosemide). AstraZeneca was one of the top ten pharmaceutical companies in the world in 2012, but faced a string of patent expiries in its blockbuster drugs.
Jenny Hantzinikolas explained the regulatory authorities that would participate in the pilot program were EMA, US FDA, MHRA (UK), ANVISA (Brazil) and PMDA/MHLW (Japan), with the scope of the pilot limited to pre-approval and pre-licensing medicine inspections, but would not include surveillance medicine inspections. Canada DHPID ).
156, as added by the 1984 Hatch-Waxman Amendments, for certain FDA-regulated products. The use of a surrogate endpoint can considerably shorten the time required prior to receiving FDA approval. If the confirmatory trial shows that the drug actually provides a clinical benefit, then the FDA grants traditional approval for the drug.
Metformin hydrochloride is a generic drug approved by the Food and Drug Administration (FDA) to help manage blood sugar levels in people with type 2 diabetes mellitus. OTC substitutes for metformin There are no exact OTC substitutes for metformin, and there aren’t any FDA-approved OTC medications for diabetes.
DEA has interpreted pharmacists’ corresponding responsibility as prohibiting the filling of a prescription where the pharmacist or pharmacy “knows or has reason to know” that the prescription is invalid. Holiday CVS, L.L.C. d/b/a CVS/Pharmacy, Nos. 219 and 5195; Decision and Order , 77 Fed. 62,316, 62,342 (Oct.
2001– FDA green lights revolutionary treatments. Just over a decade after it was developed by biochemist Nicholas Lyndon, Imatinib received US Food and Drug Administration (FDA) approval in 2001. 2012 – The 100,000 Genomics Project begins. 2002 – Emergence of CAR-T therapy.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.











































Let's personalize your content