This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Koblitz Integral to the careful balance Congress struck when passing the Hatch-Waxman Amendments, the patent term extension (PTE) is intended to restore patent life that was consumed during regulatory review of an FDA-regulated product. FDA has already been there , of course, with respect to 180-day exclusivity.)
Iloprost had also received Priority Review and Orphan Drug Designations for this indication, and FDA approval in 2004 for the treatment of pulmonary arterial hypertension.
Celgene later submitted study data to the Food and Drug Administration in its application for regulatory approval, but did not simultaneously seek additional patents on these other forms for another five years — in 2004. Patent & Trademark Office granted two more patents, but did not do so until 2008.
Between 2004 and 2021, a U.S. Of those 36 cases, two-thirds involved 34 patents that covered medical products regulated by the FDA. Food and Drug Administration were more frequently invalidated due to information misrepresented or withheld from patent examiners than any other industry sector, according to a new analysis.
In the US, botanical dietary supplements can be sold without US Food and Drug Administration (FDA) approval, 1 prompting many vendors to promote the use of botanical products as dietary supplements , 2 rather than pursue a path of regulatory approval. 3 Is two too few? 3 Is two too few?
FTC, deep in its foray into the Orange Book, filed an Amicus Brief in the case arguing that the patents do not claim any FDA-approved drug. Citing to a 2004 Supreme Court case, Verizon Commc’ns., of Trinko, LLP , 540 U.S.
Novartis said the decision to revoke the authorisation was based on a review of crizanlizumab under Article 20 of Regulation (EC) No 726/2004. Crizanlizumab is approved for use by the United States Food and Drug Administration (FDA). Novartis continues to discuss the STAND results with the FDA and other health authorities.
Food and Drug Administration (FDA) issued two guidance documents, one draft and one final, on food allergen labeling requirements. In addition to providing additional examples of tree nuts, the draft guidance states that FDA considers the following categories of fish to be major food allergens under Section 201(qq): Jawless fish (e.g.,
Bob came to ISPE in 2004, furthering an already distinguished career in the pharmaceutical industry and as a regulator. After having worked in industry as a senior Quality Assurance position he joined the Therapeutic Goods Administration (TGA), Australia in 1971 as a GMP inspector.
The assessment report of the Committee for Medicinal Products for Human Use (CHMP)’s Article 5(3) of Regulation (EC) No 726/2004 opinion on nitrosamine impurities in human medicinal products offers guidance and recommendations on mitigation and prevention of nitrosamine-contaminated human medicinal products.
Shares in US biotech Y-mAbs Therapeutics have lost almost a third of their value after FDA advisors unanimously rejected its brain cancer therapy 131I-omburtamab in 16 to 0 vote. Changes in standard and supportive care over the time period could also have skewed the results, said the FDA reviewer.
Hatch Foundation’s For cheap generic drug prices, you can thank 40 years of Hatch-Waxman We Work For Health’s Hatch-Waxman Act: Celebrating 40 Years of a Balanced and Innovative Drug Ecosystem Tradeoffs’ Race to the Bottom Series FDA CDER Conversations – 40th Anniversary of the Generic Drug Approval Pathway The U.S. Waxman (sadly, Sen.
Food and Drug Administration (FDA) released a draft update to its Compliance Policy Guide (CPG) for FDA staff on the Agency’s enforcement of major food allergen labeling and cross-contact. The draft CPG directs FDA field staff to examine possible food product adulteration due to labeling related to allergen cross-contact.
These teams will include experts in regulatory, GMP compliance and quality systems, often including ex-FDA and MHRA inspectors and industry experts. In addition, she worked as head of inspectorate and licensing for the MHRA from 2004-2006. She joined NSF in 2017.
Houck — Administrator Anne Milgram has been on the receiving end of high-level support for and against rescheduling marijuana since the Food and Drug Administration (“FDA”) and Health and Human Services (“HHS”) recommended the Drug Enforcement Administration (“DEA”) reschedule from schedule I to schedule III last August. By Larry K.
Clinical research in VR as a treatment for PTSD was accelerated in 2004 when an epidemiological study of PTSD was published , noting the rise in cases among veterans of the conflicts in Iraq and Afghanistan. Skip says this paper was a “call to arms” to find improved treatment methods.
This session promises an insightful exploration of critical topics such as EU Annex 1 Medicinal Products Rev 4, FDA CGMP 2004, and ISPE Sterile Products 3rd Edition Baseline Guide. Ryan Waldhardt brings his extensive experience at Grand River Aseptic Manufacturing to discuss the USA Perspective of the current regulatory framework.
The US Food and Drug Administration (FDA) subsequently developed a liquid chromatography – high resolution mass spectroscopy (LC-HRMS) method for the determination of NDMA in ranitidine. FDA assigned a provisional AI of 26.5 10 It is not clear whether FDA will adopt a similar t-AI. This t-AI would be used for ≤12 months.
In 2020, the Federal Trade Commission issued a report regarding settlements reached between brand and generic manufacturers in FY 2017 and noted that “for the first time since [FY] 2004, no settlement agreement in [FY] 2017 contains a no-[authorized generic] AG commitment.”. Throughout 2022, Novartis was at the opposing ends of legal cases.
Because anabolic steroids are also abused to enhance athletic performance and increase muscle strength, Congress has enacted three laws regulating anabolic steroids: the Anabolic Steroid Control Acts of 1990 and 2004, and the Designer Anabolic Steroid Control Act of 2014 (“DASCA”).
The European Medicines Agency (EMA) advocates 18 ng/day 6 as the default AI, where the US Food and Drug Administration (FDA) supports a value of 26.5 8 As such, detection of an API nitrosamine with an MDI of 4000 mg/day may be feasible for FDA submissions, but probably not with the EMA. ng/day for novel API nitrosamines. 25 June 2020.
In 2004, the Food and Drug Administration (FDA) noted that while the research is not conclusive , consuming EPA and DHA omega-3 fatty acids may help reduce the risk of heart disease. Consuming omega-3 fatty acids was shown to be beneficial in reducing the risks of heart attack and stroke.” “The
The US Food and Drug Administration (FDA) requires that manufacturers establish whether nitrosamines, including NDSRIs, could be present in active pharmaceutical ingredients (APIs) and drug products, using the “three-step mitigation strategy described in the agency’s guidance”. Nitrosamine Imp -Q&A-Mar-22.pdf. Accessed on 02 April 2022.
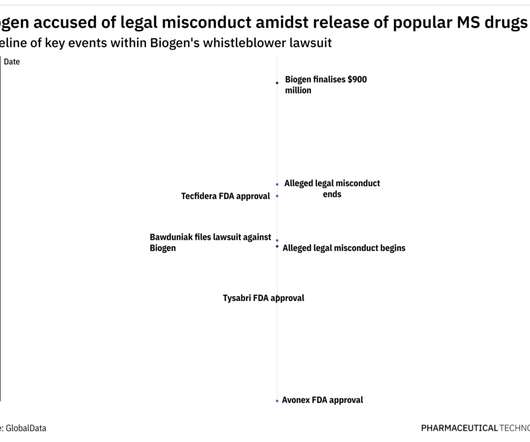
In 2009, Tysabri was performing well, having yielded $776 million in sales after being first approved in 2004, as per Biogen’s 2009 financial filings. Tecfidera was approved in 2013 to treat MS, and in August 2020, Mylan launched its first FDA-approved dimethyl fumarate generic to the market.
Hyaluronic acid injections, or rooster comb injections as they are often called, are only approved by the Food and Drug Administration (FDA) for use in osteoarthritis of the knee joint. Synvisc is one of the brand-name hyaluronic acid products indicated by the FDA for knee osteoarthritis. Who can benefit from rooster comb injections?
Based on these promising results, the US Food and Drug Administration (FDA) granted Breakthrough Therapy Designation to AlphaMedix in February 2024. He then worked at BCG from 2004 to 2011, where he developed expertise in the energy, engineering, and high-tech sectors.
A lack of transparency The importance of setting the primary outcome prior to commencing a study and not deviating from the original protocol, was first highlighted in 1990 by Jay Siegel , a physician and research scientist working for the FDA in the US. This suspicion was confirmed in a 2004 analysis by researchers from Oxford.
Since 2004, Roche/Genentech has been implementing carbon dioxide reduction measures that have led to a 59% absolute reduction in greenhouse gas (GHG) emissions from our own operations and purchased energy (Scope 1 and 2). September 2004. Now the implementation can focus on risk controls. 5 US Food and Drug Administration.
Food and Drug Administration (FDA) in 2004 , Cymbalta has become a widely used antidepressant to treat various mental health and pain-related conditions. Cymbalta may have several off-label uses in addition to its FDA-approved uses. It is also approved for fibromyalgia and chronic muscle pain. Approved by the U.S.
m/s recommendation made its way into the US Food and Drug Administration (FDA)’s “Sterile Drug Products Produced by Aseptic Processing—Current Good Manufacturing Practice” 7 and into the EC GMP Annex 1 “Manufacture of Sterile Medicinal Products” in 2003. October 2004. 5 , 6 From his research, the 0.45 8 When an air velocity of 0.45
21 Similarly, the FDA Center for Drug Evaluation and Research (CDER) began issuing electronic CPPs (eCPP) starting December 2021. April 2004. 012166 from 2004. Modernization of the FDA CDER Export Certificate Program.” The WHO endorsed the EMA’s decision and encouraged the various boards of health to accept this approach.
Why, then, are FDA inspections abroad so infrequent and disappointing? FDA Inspections Suspended: During the COVID pandemic, the Food and Drug Administration suspended its inspections at most foreign pharmaceutical facilities. Even under the best of conditions, the FDA never inspects all foreign pharmaceutical plants annually.
Published September 2004. These media fill process simulations also serve to evaluate the aseptic technique used during operator interventions. link] 3 US Food and Drug Administration. Guidance for Industry. Sterile Drug Products Produced by Aseptic Processing—Current Good Manufacturing Practice.” link] 5 Polzer, A., Koeth, and S.
During 2023, there were five revisions to the European Medicines Agency ’s (EMA’s) Questions and Answers (Q&As) for marketing authorisation holders/applicants on the CHMP Opinion for the Article 5(3) of Regulation (EC) No 726/2004 referral on nitrosamine impurities in human medicinal products. These changes are summarised in Table 1.
In humans, metronidazole is FDA-approved for treating certain bacterial infections. Although not FDA-approved for animals, metronidazole is used off-label in cats when prescribed in veterinary medicine. What is metronidazole used for in cats? The antibiotic also treats giardia infections.
The latest revelation continues a troubled spell for Mesoblast, which announced the Novartis alliance shortly after the FDA rejected remestemcel-L – under the Ryoncil brand name – as a treatment for children with graft versus host disease (GvHD). It has made cumulative loses of almost $650 million since being founded in 2004.
2 The US Food and Drug Administration (FDA) has recently announced that it will control the quality of tobacco products, particularly e-cigarettes, more closely, to prevent avoidable contamination and help address “inconsistencies between product labelling and actual concentrations” in these products, potentially misleading customers.
Three SMN-enhancing treatments for the condition are approved by the US Food and Drug Administration (FDA) 2 , however there is no cure for SMA at present. While Zolgensma is also being investigated in the form of an injection, the MOA currently approved by the FDA is a single infusion.
In 2004, the U.S. Food and Drug Administration (FDA) began requiring a “ black box warning ” on antidepressants. An increase in suicidal thoughts New or increasing suicidal thoughts or behaviors can indicate that your antidepressant dose is too high.
This year, clascoterone (brand name: Winlevi) has been approved by The United States Food and Drug Administration (FDA) for treatment of acne in the US. In the UK, there are several options for the management of acne, including topical preparations and oral antibiotics. Sebum’s role is to lubricate the hair and skin.
Market Cap: $8.46M Founded Year: 2004 Total Employees: ~4 Headquarters: Texas, United States Stock Exchange: OTCMKTS QSam Biosciences is a clinical-stage biotech company focused on developing and commercializing targeted therapeutic radiopharmaceutical products for treating cancer and related diseases.
Recent US FDA data show that the lack of raw material availability contributes to 27% of drug shortages (see Appendix ). It is considered a moderate change by the FDA (CBE30) and NMPA. This change ranges from a moderate change (CBE-30) by the FDA to a minor change not requiring prior approval by the WHO.
Fortunately, my colleague Dr. Kirk Gair from West Covina, CA, who is also a Hashimoto’s patient, has used cold lasers in his clinic since 2004 and has developed protocols that combine LLLT with chiropractic modalities. This Class II medical device is FDA-cleared and meant to target all kinds of pain.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.














































Let's personalize your content