This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Food and Drug Administration (FDA) issued two guidance documents, one draft and one final, on food allergen labeling requirements. In addition to providing additional examples of tree nuts, the draft guidance states that FDA considers the following categories of fish to be major food allergens under Section 201(qq): Jawless fish (e.g.,
In the US, botanical dietary supplements can be sold without US Food and Drug Administration (FDA) approval, 1 prompting many vendors to promote the use of botanical products as dietary supplements , 2 rather than pursue a path of regulatory approval. 3 Is two too few? 3 Is two too few?
The assessment report of the Committee for Medicinal Products for Human Use (CHMP)’s Article 5(3) of Regulation (EC) No 726/2004 opinion on nitrosamine impurities in human medicinal products offers guidance and recommendations on mitigation and prevention of nitrosamine-contaminated human medicinal products.
Bob came to ISPE in 2004, furthering an already distinguished career in the pharmaceutical industry and as a regulator. Bob also initiated an agreement under which ISPE’s Guidance Documents are made available to PIC/S and WHO inspectors, which is still in place today.
Why, then, are FDA inspections abroad so infrequent and disappointing? FDA Inspections Suspended: During the COVID pandemic, the Food and Drug Administration suspended its inspections at most foreign pharmaceutical facilities. Even under the best of conditions, the FDA never inspects all foreign pharmaceutical plants annually.
Shares in US biotech Y-mAbs Therapeutics have lost almost a third of their value after FDA advisors unanimously rejected its brain cancer therapy 131I-omburtamab in 16 to 0 vote. Changes in standard and supportive care over the time period could also have skewed the results, said the FDA reviewer.
These teams will include experts in regulatory, GMP compliance and quality systems, often including ex-FDA and MHRA inspectors and industry experts. In addition, she worked as head of inspectorate and licensing for the MHRA from 2004-2006. She joined NSF in 2017.
This track will delve into five important topics where you will hear from speakers who will help answer your questions and provide you with the necessary information to make confident decisions regarding any updated changes to your facility, equipment, procedures, and documents.
For example, an assessment for a facility primarily focused on US and EU markets would only consider GMPs and regulations from EudraLex and the FDA. EU human tissues and cells directive 2004/23/EC; 7. EU human tissues and cells directive 2004/23/EC; 7. FDA CFR Title 21 Parts 211, 600, and 1271; 8. ,
A lack of transparency The importance of setting the primary outcome prior to commencing a study and not deviating from the original protocol, was first highlighted in 1990 by Jay Siegel , a physician and research scientist working for the FDA in the US. This suspicion was confirmed in a 2004 analysis by researchers from Oxford.
Improvements have been observed by various regulatory agencies in Latin America for the acceptance and implementation of international standards—for instance, the ICH Common Technical Document (CTD) format. Additional required information beyond ICH guidelines, or non-value-added documents (e.g., 6, and 2.3.S.7 7 are required.
The US Food and Drug Administration (FDA) requires that manufacturers establish whether nitrosamines, including NDSRIs, could be present in active pharmaceutical ingredients (APIs) and drug products, using the “three-step mitigation strategy described in the agency’s guidance”. Nitrosamine Imp -Q&A-Mar-22.pdf. Accessed on 02 April 2022.
Attendees were invited to submit questions to the FDA representatives. Please note that views expressed by the panelists are not necessarily representative of the position of the FDA, and that questions and responses are lightly edited for clarity. How will the FDA use Annex 1 in its final version?
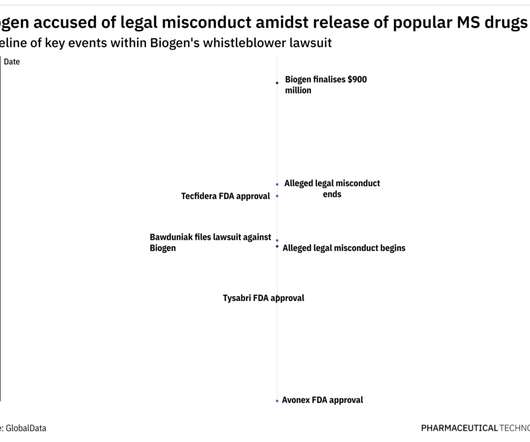
In 2009, Tysabri was performing well, having yielded $776 million in sales after being first approved in 2004, as per Biogen’s 2009 financial filings. These, along with other similar claims, meant Biogen owed potential damages of $1,036,900,151 to the US and the various States, as per court documents.
Also, determining whether an intrusion occurred and documenting that intrusion adds to the challenges of performing aseptic technique properly (and perhaps adds to the subjectivity of the current control strategies). Published September 2004. link] 3 US Food and Drug Administration. Guidance for Industry. link] 5 Polzer, A.,
Since 2004, Roche/Genentech has been implementing carbon dioxide reduction measures that have led to a 59% absolute reduction in greenhouse gas (GHG) emissions from our own operations and purchased energy (Scope 1 and 2). September 2004. Now the implementation can focus on risk controls. 5 US Food and Drug Administration.
September 2004. September 2004. FDA Official Warns Manufacturers of Common Problems Found in Aseptic Operations.” www.raps.org/news-and-articles/news-articles/2023/3/fda-official-warns-manufacturers-of-common-problem 25 US Food and Drug Administration Center for Drug Evaluation and Research. 3 (2022):593–607.
Recent US FDA data show that the lack of raw material availability contributes to 27% of drug shortages (see Appendix ). A formal change control system under the company’s pharmaceutical quality system (PQS) is required to evaluate all raw material changes, with established procedures for identification, documentation, review, and approval.
The setpoint for “proper” air velocity in cleanroom systems is documented in standards and regulations as 0.45 9 In 2015, the FDA published a guidance manual 10 that provided questions for inspections, including: Is the air flow in critical areas unidirectional when delivered to the point of use? October 2004. m/s up to 0.54
Fortunately, my colleague Dr. Kirk Gair from West Covina, CA, who is also a Hashimoto’s patient, has used cold lasers in his clinic since 2004 and has developed protocols that combine LLLT with chiropractic modalities. This Class II medical device is FDA-cleared and meant to target all kinds of pain. References [1] Nanan R, Wall JR.
The writing of requirements, design documentation, and test scripts, and the configuration of hardware and software, can be conducted from home. During the COVID-19 pandemic, the US FDA issued guidelines for remote evaluations of drug manufacturing and bioresearch facilities. This is achieved by off-the-shelf technology.
A 2015 case report documented the thyroid labs of a man who had recurrent hyperthyroid episodes after three of his wife’s pregnancies! Published 2004 Aug 18. Like women, men can develop hormonal changes after the birth of a child, which can be linked to postpartum depression. [14] His wife had Hashimoto’s.) BMC Psychiatry.
In 2020, the Federal Trade Commission issued a report regarding settlements reached between brand and generic manufacturers in FY 2017 and noted that “for the first time since [FY] 2004, no settlement agreement in [FY] 2017 contains a no-[authorized generic] AG commitment.”. Throughout 2022, Novartis was at the opposing ends of legal cases.
Questions and Answers Regarding Food Allergens, Including the Food Allergen Labeling Requirements of the Federal Food, Drug, and Cosmetic Act This final guidance replaces previous draft and final guidance documents on food allergen labeling that FDA issued in November 2022, which we discussed in a previous post. By Sophia R.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.




























Let's personalize your content